目标区域捕获联合新一代测序技术在遗传性聋研究中的应用及发展前景*
2014-06-12王洪阳综述王秋菊审校
王洪阳 综述 王秋菊 审校
耳聋是最常见的遗传异质性疾病之一,大多数遗传性聋是单基因病。自1988年第一个耳聋基因被定位,1995年第一个非综合征型耳聋基因POU3F4被克隆以来,耳聋基因的定位及克隆研究取得了巨大进展,目前已经鉴定了76个非综合征型耳聋相关基因,另外100多个已定位的耳聋基因座尚待相关基因的鉴定(截至2013年10月)[1]。在单基因病的致病基因定位和克隆研究中,通常采用的家系连锁分析和定位克隆技术是最有效、最准确的方法之一,但是,如果家系中患病人数有限或不外显,或外显不全,或是散发性病例,则连锁分析多半失效。此外,以Sanger测序为基础的测序技术因其高成本和长耗时也限制了其应用。新一代测序技术(next-generation sequencing technology, NGS)为探索单基因病提供了新的途径,因其无需收集大家系的样本,且可对全外显子组或全基因组的无偏倚测序,使得NGS在发现罕见遗传病的病因方面具有独特优势,尤其是对遗传性聋患者的全外显子组或耳聋相关基因组(分别占全基因组的1%和0.01%)[2]进行目标区域测序分析,捕获的是疾病的大部分致病突变信息,具有所需样本量少、费用低、通量高的优势,能够检测更多的样本,促进了遗传性聋新的致病变异的发现。本文聚焦于目标区域捕获联合NGS在遗传性聋基因研究中的应用,尤其是该技术在转化医学和新生儿听力筛查中的应用前景,同时归纳了基因捕获的主要方法和新一代测序技术的发展及其特点。
1 新一代测序技术
基因组是研究人类疾病的核心和基础,基因组技术的进步彻底改变了鉴定人类基因突变的途径。DNA测序技术发展的第一阶段是以Sanger测序为基础的第一代测序技术,2005年,Roche公司的454测序仪的出现标志着NGS问世,随后,Illumina公司的Solexa测序仪和ABI公司的SOLiD测序仪相继出现;不同的测序平台在PCR模式、技术原理、测序模式及读长等方面各有特点(图1),NGS也称大规模平行测序或深度测序,其低成本及高通量并行测序的优势,使疾病的研究模式发生了重大转变,但同时也具有文库构建复杂、测序长度短和错误率偏高等缺点。为解决早期新一代测序技术的问题,最近几年出现了几种新的高通量测序技术,通过改变测序原理或检测手段,实现了更加简化的测序过程,更低的测序成本和更高的测序速度:如单分子测序,能够直接观察和测定DNA或RNA分子的数量和区域结构,主要包括HeliScope公司的遗传分析系统、VisiGen公司的单分子合成测序、Pacific Biosciences公司的单分子实时测序技术以及Oxford纳米孔测序技术等,这几种新技术也被称为第三代高通量测序技术(third-generation sequencing technology, TGS)[3],三代测序技术各有其优缺点,其特点比较见表1。
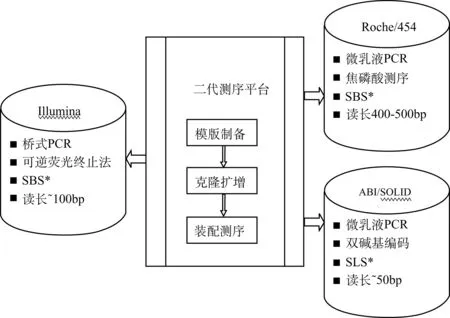
图1 新一代测序平台比较[3]

表1 第一~三代测序技术的特点比较
2 目标区域捕获技术
2.1DNA水平的测序策略 目标区域捕获联合NGS是利用定制的探针与基因组DNA进行杂交或PCR捕获感兴趣的基因组区域,然后应用高通量测序技术进行测序的策略,具有经济、精确、可行、周转时间短等优势,能够检测更多的样本。有文献报道85%的人类致病突变位于占全基因组序列1%的蛋白质编码序列,即外显子序列上[7]。针对基因组蛋白质编码区的全外显子组测序(whole exome sequencing, WES)是介于全基因组关联分析(genome-wide association study, GWAS)与全基因组测序(whole genome sequencing, WGS)之间的基因分析策略[8],利用目标区域捕获将基因组的全部外显子区域捕获后进行高通量测序,以发现疾病相关遗传变异,其本质上也是一种目标区域测序技术,可以发现基因编码区的低频变异及对疾病具有不同效应的稀有变异。与全基因组测序相比,在相同成本下,目标区域测序技术可对更多个体的蛋白编码信息进行研究,可获得覆盖度更深、准确性更高的数据。目标区域捕获技术具有简便、经济、高效的优势,可用于单基因病、复杂疾病及癌症等的致病基因或易感基因的研究[9,10]。过去两年中,WES已鉴定了1 000多种罕见单基因病的基因[10]。
2.2不同目标区域捕获技术的比较 传统的靶向富集技术是PCR和杂交,现今的靶向富集技术可以分为固相捕获、液相捕获、分子倒位探针(molecular inversion probes, MIP)和Spacer Multiplex Amplification Reac Tion(SMART)(表2),其中固相捕获及液相捕获是以杂交为基础的富集技术,而MIP和SMART是以PCR为基础的非杂交富集技术,后两者需要特殊的设备,不适用于高通量应用,因此只应用于特殊的基因测序;固相捕获以芯片为载体,与设计的探针序列进行耦联,进而对杂交后富集的DNA片段进行测序分析;液相捕获原理与固相芯片捕获相似,只是同探针进行耦联的是微珠而非固相载体芯片。与固相芯片相比,液相捕获具有成本低、操作简便、所需DNA量少等优势[12]。

表2 常用靶向富集技术比较[11]
注:*CTR(capture target region)为目标捕获区域
3 目标区域捕获联合NGS在遗传性聋研究中的应用
众所周知,不同民族的耳聋基因突变频率不同,目前多数耳聋基因研究局限于人类最常见的突变基因,以中国大陆人群为例,30%左右的遗传性聋患者与GJB2、SLC26A4和线粒体12SrRNA突变有关,可通过常见的耳聋基因筛查明确诊断,而另外的70%左右的遗传性聋却无法明确病因,需要进行深度挖掘[13]。因为导致耳聋的大量致病基因以及复杂突变谱的存在,以基因特异性的Sanger测序作为遗传性聋的基因诊断策略成本高、耗时长,且不同民族的常见基因突变不同也使其应用受限[2]。目标区域捕获测序为耳聋基因研究提供了新的路径,可在一次检测中包含所有已知耳聋基因,检测所有类型的突变,并可检测传统方法难以检测的大基因,同时可应用于散发病例,是目前研究遗传性聋的有效技术。2009年11月,Nature Genetics在线发表了首篇应用WES研究单基因病的论文,在3个家庭的4个米勒综合征患者中筛选出致病基因DHODH[14],首次证明了WES是快速定位单基因病致病基因的有效工具;此后,WES在单基因病研究中发挥了越来越多的重要作用,据估计WES可发现约60%单基因病的致病基因[15]。
3.1目标区域测序在遗传性聋研究中的应用实例 目标区域捕获联合新一代测序途径最近也鉴定出新的遗传性聋基因,这些研究表明针对目标区域或全外显子组的NGS,结合在非同源家系中验证、功能和免疫标记检测技术,可以在小家系中鉴定致病基因。最先应用这一策略的是Rehman等[16],利用NimbleGen固相捕获芯片技术,结合Roche 454测序仪鉴定了导致非综合征型聋的致病基因C9orf75(之后重命名为TPRN);首先捕获了DFNB79位于染色体9q34.3(包含108个候选基因)的目标区域,然后对该2.9 Mbp的区域进行测序,在一个近亲家系中患者的目标区域检测到8个尚未报道的纯合变异,其中6个是多态性表现,1个位于非编码区,另外1个是位于所预测基因C9orf75上的一个无义突变;此后在针对DFNB79患者的检测中发现了该基因的3个移码突变,免疫标记也显示小鼠耳蜗中TPRN编码蛋白高表达。此后,特定区域捕获或全外显子组捕获技术成功应用于多型耳聋分子基础的研究(表3)。近两年,目标区域捕获联合NGS开始应用于小规模人群研究,为建立耳聋分子流行病学数据库及筛选热点突变组合奠定了基础;Shearer等[38]对100例非综合征型遗传性聋样本进行捕获测序,涵盖了67个已知耳聋基因990个基因变异,诊断率为42%;杨涛等[39]对125个排除了常见致病突变的耳聋样本进行捕获测序,捕获区域包括79个已知耳聋基因,最后在15个非常见基因上鉴定了28个致病突变,诊断率为17.4%。小规模人群研究一方面说明已知耳聋基因捕获测序与常见耳聋基因筛查相比,提高了耳聋基因诊断率,另一方面也说明耳聋基因研究任重而道远,仍有一半以上的遗传性聋病因不明,有待进一步的研究。
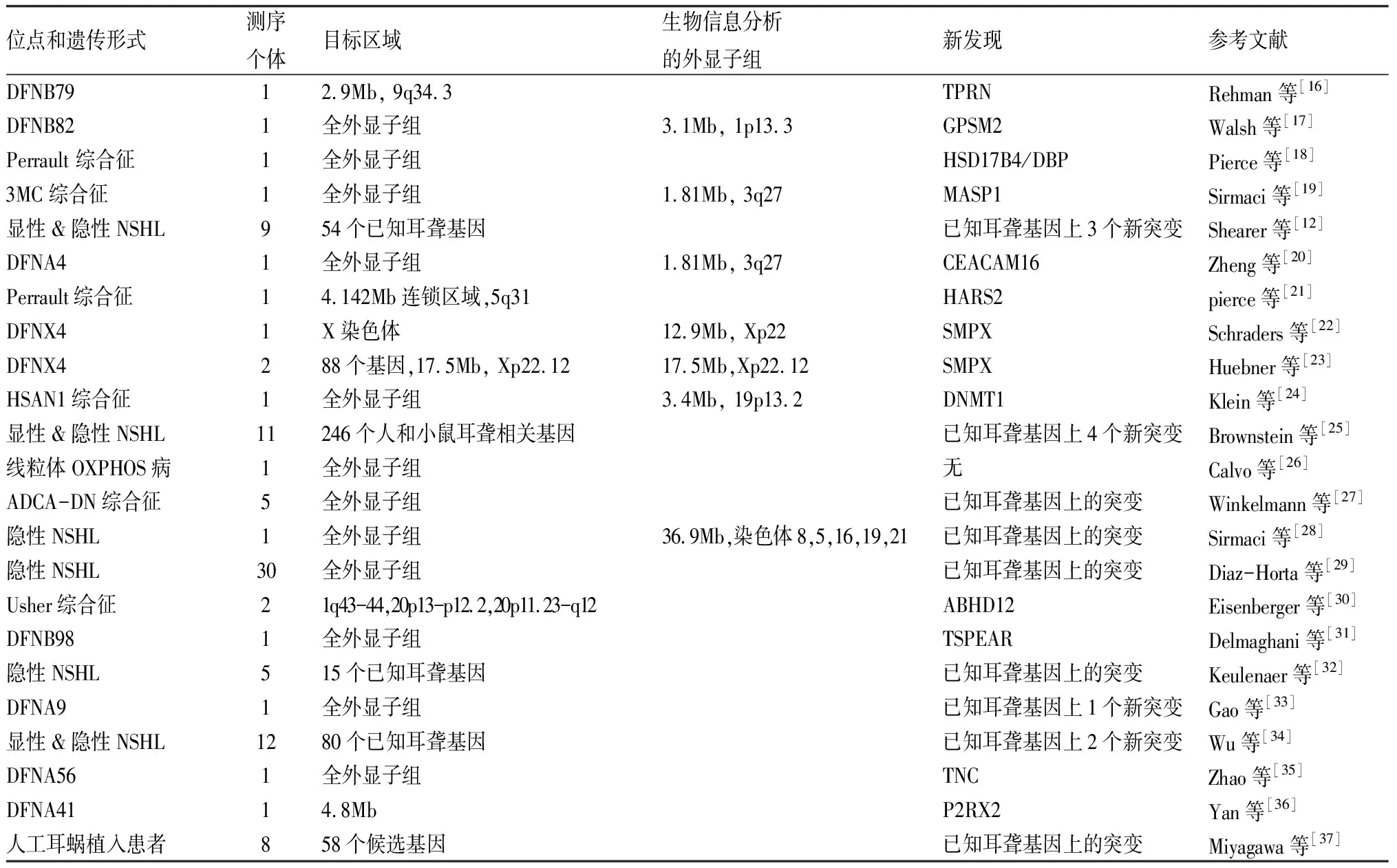
表3 目标区域捕获联合新一代测序技术在听力研究中的应用
3.2目标区域测序可行性的验证 最近的一些研究从多方面验证了NGS检测遗传性聋的可行性(表4)。Shearer等[12]评价了针对54个已知的非综合征型耳聋基因进行靶向富集联合新一代测序技术的可行性,目标基因的捕获同时使用了固相捕获和液相捕获,并对Sureselect-Illumina和NimbleGen-454两个平台进行了比较,结果在6个特发性聋样本中鉴定了5个致病突变,对目标区域测序在耳聋遗传诊断方面的敏感度、特异度和可重复性给予了肯定。Brownstein等[25]应用的是包含246个耳聋相关基因的cRNA寡核苷酸探针(82个人类蛋白编码基因,2个人miRNA和162个小鼠耳聋基因中人的同源基因),从11个先证者的外周血中获得配对末端库,应用Illumina的NGS平台进行测序,结果在6例先证者中发现了致病突变,分别位于CDH23、MYO15A、TECTA、TMC1和WFS1,并在另外的20例先证者及其家系中鉴定了致病等位基因;同时,他们发现了摩洛哥犹太人群中TMC1上的一个始祖等位基因突变,仅该隐性突变TMC1(p.S647P)就可解释摩洛哥犹太人群中34%的遗传性聋。在针对中国人的目标区域测序中,Wei等[40]设计了包含69个耳聋相关基因的芯片,该芯片首次加入了线粒体基因,然后对携带有5个已知突变的10个患者进行目标区域测序,验证了该方法的可行性及应用价值,随后在一个耳聋家系中鉴定了基因MYO7A上的复合杂合突变。尽管可行性多次得到验证且硕果累累,但捕获目标外显子的高昂费用仍是限制NGS在人群中大规模应用的主要阻碍。新的捕获测序策略不断出现,例如Tang等[42]设计了一个cDNA探针来进行目标基因的富集以降低费用,Keulenaer等应用半自动PCR扩增联合NGS,克服了以杂交为基础的目标区域捕获敏感性差的缺点[32]。
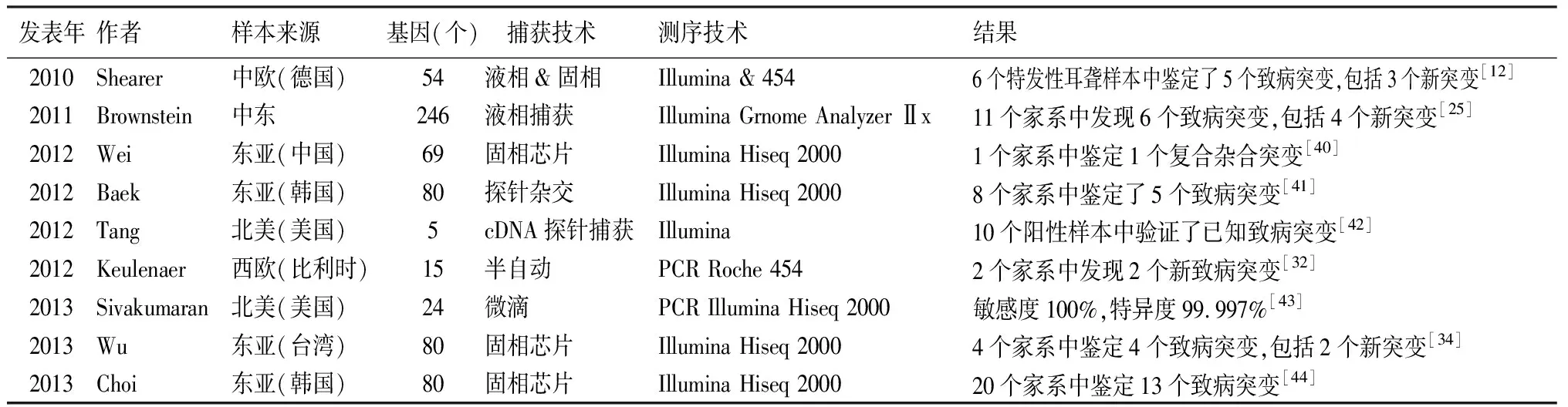
表4 近3年验证NGS检测耳聋基因诊断效用的文献
3.3面临的挑战 NGS途径产生的突变结果仍需Sanger测序进行验证,数据质控缺乏管理监督,数据分析及数据报道的无统一标准均限制了它的快速发展[45]。如何使不同的NGS平台的数据标准化以便进行比较仍然悬而未决[46],而因其测序时重复区域的碱基延伸的不可靠性而导致的错误率偏高问题亦亟待解决[47],样品准备和文库构建尚待简化,海量数据的分类、存档、分析仍是难题。技术层面之外,伦理问题不容小视,从NGS海量数据中可以推测出许多属个人隐私信息,甚至是亲属及相关民族的信息,尽管各单位伦理委员会对患者信息和数据的使用和储存有严格的规定,目前仍缺乏对NGS数据使用管理的专一规则。此外,外显子组测序技术的固有缺陷使其只能捕获外显子区域内部和边界的变异信息,对大部分发生在非编码区的变异无能为力,且不能检测到基因组内较大的结构变异和基因拷贝数变化。
3.4应用前景 NGS的应用正在进入临床实践,临床医生有必要掌握医学遗传学的知识,以便更有效地进行疾病的诊断、治疗、咨询和预防。然而对数据的解释分析及将探索结果转化为临床应用仍然充满挑战,目前最迫切的任务是从个人基因组上百万的变异中筛选出致病突变。另一个很重要的方面是需同时提高遗传咨询能力,以解释NGS产生的海量数据。不久的将来,临床医生有可能通过综合分析既往史、家族史和NGS数据,鉴定有疾病倾向的变异以及个体中影响药物代谢的变异,遗传性疾病携带者也可能成为患者既往史的一部分。基因组学的发展很有可能最终导致遗传医学的诞生,推进人类健康的重大进步和发展[48]。
另一可能的应用方向是新生儿耳聋基因筛查。2000年我国开始在全国范围内开展新生儿听力筛查工作,新生儿听力筛查对耳聋的早期预防和干预意义重大,然而传统的听力筛查有假阳性率高、不能确定耳聋的潜在病因、不能发现迟发性聋以及随访率低等缺点。大多数先天性和儿童早期发病的耳聋是基因突变导致的,耳聋遗传信息对于疾病的诊断、治疗及遗传咨询有重大意义。新生儿听力与基因联合筛查具有指导抗生素应用、指导部分耳聋患者减缓耳聋的发展、预测人工耳蜗植入的效果以及进行遗传咨询、评价再次生育子女出现耳聋的几率等意义[49]。随着目标区域捕获和NGS的发展,可以通过一次实验,检测所有已知和候选的耳聋基因[25],从而使具有高度遗传异质性耳聋的DNA诊断取得重大进展。
此外,随着临床转化的逐步成熟,目标区域NGS还有望应用于高危人群的检查、聋哑患者生育风险检测及评估以利于优生优育。新一代测序平台的高灵敏性,可对孕妇血浆中存在的微量胎儿DNA进行检测,可针对包括遗传性聋在内的100种常见疾病的常见基因进行捕获测序,实现无创性产前筛查及对遗传性疾病的分析。
4 总结和展望
高通量及低成本的NGS极大地促进了遗传性聋基因组学的研究进展,其中包括全外显子组捕获在内的目标区域捕获联合新一代测序技术在遗传性聋研究中具有广泛的应用前景,作为全基因组测序的重要补充,研究者可以根据研究需要选择合适的基因测序策略,从而更加经济高效地完成基因测序目的。然而,该技术的临床应用仍然充满挑战,数据的分析管理等诸多问题有待解决,生物学信息的发展及管理制度的完善将有助于该技术更好地应用。此外,基因组水平研究是整个疾病研究的基础,为了全面深入揭示遗传性疾病的发生发展规律及遗传特点,除基因组水平研究外,基于新一代测序技术的转录组、蛋白组及表观组、宏基因组的系统研究是未来遗传性疾病的研究趋势,进而更好地为临床筛查以及诊治遗传性疾病提供思路。
5 参考文献
1 Van Gamp G, Smith RJH. Hereditary hearing loss homepage, 2013. [http://hereditaryhearingloss.org].
2 Lin X, Tang WX, Ahmad S, et al. Application of targeted gene capture and next-generation sequencing technologies in studies of human deafness and other genetic disabilities[J]. Hearing Res, 2012, 288: 67.
3 Chandra SP, Rafal S, Andrzej T. Sequencing technologies and genome sequencing[J]. J Appl Genetics, 2011, 52: 413.
4 Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome[J]. Nature, 2001, 409: 860.
5 Wheeler DA, Srinivasan M, Egholm M, et al. The complete genome of an individual by massively parallel DNA sequencing[J]. Nature, 2008, 452: 872.
6 Clarke J, Wu HC, Jayasinghe L, et al. Continuous base identification for single-molecule nanopore DNA sequencing[J]. Nat Nanotechnol, 2009, 4:265.
7 Stenson PD, Mort M, Ball EV, et al. The human gene mutation database: 2008 update[J]. Genome Med, 2009, 1: 13.
8 Mardis ER. A decade's perspective on DNA sequencing technology[J]. Nature, 2011, 470: 198.
9 Brownstein Z, Bhonker Y, Karen BA, et al. High-throughput sequencing to decipher the genetic heterogeneity of deafness[J]. Genome Biol, 2012, 13: 245.
10 Rabbani B, Mahdieh N, Hosomichi K, et al. Next-generation sequencing: impact of exome sequencing in characterizing mendelian disorders[J]. Hum Genet, 2012, 57: 621.
11 Mamanova L, Coffey AJ, Scott CE, et al. Targeted-enrichment strategies for next-generation sequencing[J]. Nat Methods, 2010, 7: 111.
12 Shearer AE, Deluca AP, Hildebrand MS, et al. Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing[J]. Proc Natl Acad sci USA, 2010, 107: 21104.
13 Wang QJ. Hereditary hearing impairment will face opportunity and challenge from clinic to base[J]. Zhonghua Yi Xue Za Zhi, 2009, 89: 2809.
14 Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a mendelian disorder[J]. Nat Genet, 2010, 42: 30.
15 Gilissen C, Hoischen A, Brunner HG, et al. Disease gene identification strategies for exome sequencing[J]. Eur J Hum Genet, 2012, 20: 490.
16 Rehman AU, Morell RJ, Belyantseva IA, et al. Targeted capture and next-generation sequencing identifies C9orf75, encoding taperin, as the mutated gene in nonsyndromic deafness DFNB79[J]. Am J Hum Genet, 2010, 86: 378.
17 Walsh T, Kanaan M, Elkan-Miller T,et al. Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82[J]. Am J Hum Genet, 2010, 87: 90.
18 Pierce SB, Walsh T, Chisholm KM, et al. Mutations in the DBP-deficiency protein HSD17B4 cause ovarian dysgenesis, hearing loss, and ataxia of Perrault Syndrom[J]. Am J Hum Genet, 2010, 87: 282.
19 Sirmaci A, Walsh T, Akay H, et al. MASP1 mutations in patients with facial, umbilical, coccygeal, and auditory findings of Carnevale, Malpuech, OSA, and Michels syndrom[J]. Am J Hum Genet, 2010, 87: 679.
20 Zheng J, Miller KK, Yang T, et al. Carcinoembryonic antigen-related cell adhesion molecule 16 interacts with a-tectorin and is mutated in autosomal dominant hearing loss(DFNA4) [J]. Proc Natl Acad Sci USA, 2011, 108: 4218.
21 Pierce SB, Chisholm KM, Lynch ED, et al. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome[J]. Proc Natl Acad Sci USA, 2011, 108: 6543.
22 Schraders M, Haas SA, Weegerink NJ, et al. Next-generation sequencing identifies mutations of SMPX, which encodes the small muscle protein, X-linked, as a cause of progressive hearing impairment[J]. Am J Hum Genet, 2011, 88: 628.
23 Huebner AK, Gandia M, Frommolt P, et al. Nonsense mutations in SMPX, encoding a protein responsive to physical force, result in X-chromosomal hearing loss[J]. Am J Hum Genet, 2011, 88: 621.
24 Klein CJ, Botuyan Mv, Wu Y, et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss[J]. Nat Genet, 2011, 43: 595.
25 Brownstein Z, Friedman LM, Shahin H, et al. Targeted genomic capture and massively parallel sequencing to identify genes for hereditary hearing loss in middle eastern families[J]. Genome Biol, 2011, 12: R89.
26 Calvo SE, Compton AG, Hershman SG, et al. Molecular diagnosis of infantile mitochondrial disease with targeted next-generation sequencing[J]. Sci Transl Med, 2012, 4: 118ra10.
27 Winkelmann J, Lin L, Schormair B, et al. Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy[J]. Hum Mol Genet, 2012, 21: 2205.
28 Sirmaci A, Edwards YJ, Akay H, et al. Challenges in whole exome sequencing: an example from hereditary deafness[J]. PLoS One, 2012, 7: e32000.
29 Diaz-Horta O, Duman D, Foster J ll, et al. Whole-exome sequencing efficiently detect rare mutations in autosomal recessive nonsyndromic hearing loss[J]. PLoS One, 2012, 7: e50628.
30 Eisenberger T, Slim R, Mansour M, et al. Targeted next-generation sequencing identifies a Homozygous nonsense mutation in ABHD12, the gene underlying PHARC, in a family clinically diagnosed with Usher Syndrome type3[J]. Orphanet J of Rare Disease, 2012, 7: 59.
31 Delmaghani S, Aghaie A, Michalski N, et al. Defect in the gene encoding the EAR/EPTP domain-containing protein TSPEAR causes DFNB98 profound deafness[J]. Hum Mol Genet, 2012, 21:3835.
32 De Keulenaer S, Hellemans J, Lefever S, et al. Molecular diagnostics for congenital hearing loss including 15 deafness genes using a next generation sequencing platform[J]. Med Genomics, 2012, 5:17.
33 Gao J, Xue J, Chen L, et al. Whole exome sequencing identifies a novel DFNA9 mutation, C162y[J]. Clin Genet, 2013, 83: 477.
34 Wu CC, Lin YH, Chen PJ, et al. Application of massively parallel sequencing to genetic diagnosis in Multiplex families with idiopathic sensorineural hearing impairment[J]. PLoS One, 2013: 8: e57369.
35 Zhao YL, Zhao FF, Zong L, et al. Exome sequencing and linkage analysis identified tenascin-C (TNC) as a novel causative gene in nonsyndromic hearing loss[J]. PLoS One, 2013, 8:e69549.
36 Yan D, Zhu Y, Walsh T, et al. Mutation of the ATP-gated P2X(2) receptor leads to progressive hearing loss and increased susceptibility to noise[J]. Proc Natl Acad Sci, 2013, 110: 2228.
37 Miyagawa M, Nishio SY, Ikeda T, et al. Massively parallel DNA sequencing successfully identifies new causative mutations in deafness genes in patients with cochlear implantation and EAS[J]. PLoS One, 2013, 8: e75793.
38 Shearer AE, Black-Ziegelbein EA, Hildebrand MS, et al. Advancing genetic testing for deafness with genomic technology[J]. J Med Genet, 2013, 50:627.
39 Yang T, Wei XM, Chai YC, et al. Genetic etiology study of the non-syndromic deafness in Chinese Hans by targeted next-generation sequencing[J]. Orphanet J of Rare Dis, 2013, 8:85.
40 Wei XM, Sun YY, Yi X, et al. Next-generation sequencing identifies a novel compound heterozygous mutation in MYO7A in a Chinese patient with Usher Syndrome 1B[J]. Clinica Chmica Acta, 2012, 413:1866.
41 Baek JI, Oh SK, Kim DB, et al. Targeted massive parallel sequencing: the effective detection of novel causative mutations associated with hearing loss in small families[J]. Orphanet J of Rare Diseases, 2012, 7: 60.
42 Tang WS, Qian D, Lin X, et al. A low-cost exon capture method suitable for large-scale screening of genetic deafness by the massively-parallel sequencing approach[J]. Genet Test Mol Biomarkers, 2012, 16: 536.
43 Sivakumaran TA, Husami A, Kissell D, et al. Performance evaluation of the next-generation sequencing approach for molecular diagnosis of hereditary hearing loss[J]. Otolaryngol Head Neck Surg, 2013 [Epub ahead of print]
44 Choi BY, Park G, Gim J, et al. Diagnostic application of targeted resequencing for familial nonsyndromic hearing loss[J]. PLoS One, 2013, 8: e68692.
45 Bunnik EM, Schermer MH, Janssens C. Personal genome testing: test characteristics to clarify the discourse on ethical, legal and societal issues[J]. BMC Med Ethics, 2011, 12:11.
46 Nothnagel M, Herrmann A, Wolf A, et al. Technology-specific error signatures in the 1000 Genomes Project data[J]. Hum Genet, 2011, 130:505.
47 Nakamura K, Oshima T, Morimoto T, et al. Sequence-specific error profile of Illumina sequencers[J]. Nucleic Acids Res, 2011, 39: e90.
48 Gonzalez-Angulo AM, Hennessy BT, Mills GB. Future of personalized medicine in oncology: a systems biology approach[J]. J Clin Oncol, 2010, 28: 2777.
49 Wang QJ, Zhao YL, Rao SQ, et al. Newborn hearing concurrent gene screening can improve care for hearing loss: a study on 14,913 Chinese newborns[J]. Int J Pediatr Otorhi, 2011, 75: 535.
