利用可重复使用的URA3标记基因建立热带假丝酵母基因敲除系统
2014-05-25项峥陈献忠张利华沈微樊游陆茂林
项峥,陈献忠,张利华,沈微,樊游,陆茂林
1. 江南大学生物工程学院,工业生物技术教育部重点实验室,无锡 214122;
2. 江苏省微生物研究所有限责任公司,无锡 214063
利用可重复使用的URA3标记基因建立热带假丝酵母基因敲除系统
项峥1,陈献忠1,张利华1,沈微1,樊游1,陆茂林2
1. 江南大学生物工程学院,工业生物技术教育部重点实验室,无锡 214122;
2. 江苏省微生物研究所有限责任公司,无锡 214063
热带假丝酵母(Candida tropicalis)在发酵工业中具有重要的应用潜力,但二倍体遗传结构和较低的遗传转化效率限制了其代谢工程育种技术的应用。建立可靠的遗传转化技术并高效的删除目的基因是代谢工程改造热带假丝酵母的重要前提。文章以C. tropicalis ATCC 20336为出发菌株,通过化学诱变筛选获得了尿嘧啶缺陷型突变株C. tropicalis XZX(ura3/ura3)。以丙酮酸脱羧酶(Pyruvate decarboxylase,PDC)基因作为靶基因构建了两端包含同源臂并在选择性标记C. tropicalis URA3(Orotidine-5′-phosphate decarboxylase,乳清酸核苷-5-磷酸脱羧酶)基因两侧同向插入源于沙门氏菌(Salmonella typhimurium)的hisG序列的基因敲除盒PDC1-hisG-URA3-hisGPDC1(PHUHP),并转化宿主菌株C. tropicalis XZX,筛选获得PHUHP片段正确整合到染色体的PDC基因位点的转化子XZX02。在此基础上,将转化子 XZX02涂布于5-FOA(5-氟乳清酸)选择培养基上,筛选得到URA3基因从PHUHP片段中丢失的营养缺陷型菌株XZX03。进一步构建了第2个PDC等位基因的删除表达盒PDCm-URA3-PDCm,并转化C. tropicalis XZX03菌株,获得转化子C. tropicalis XZX04。经PCR和DNA测序确认转化子C. tropicalis XZX04细胞染色体上的两个PDC等位基因被成功敲除。文章建立了一种营养缺陷型标记可重复使用的热带假丝酵母遗传转化技术,利用该技术成功敲除了细胞的 PDC基因,为进一步利用代谢工程改造热带假丝酵母奠定了基础。
热带假丝酵母;遗传转化;同源重组;URA3基因;基因敲除
热带假丝酵母(Candida tropicalis)是一种二倍体酵母,在发酵工业中有着广泛的应用。作为一种重要的工业微生物,热带假丝酵母胞内高效的 β-氧化途径赋予其能够利用烷烃和脂肪酸作为唯一碳源,合成长链二元酸。目前,利用热带假丝酵母发酵法生产长链二元酸已应用于工业生产中[1]。热带假丝酵母也能转化木糖生产木糖醇。木糖醇在食品、医药等领域具有重要的应用。传统的化学合成方法生产木糖醇工艺复杂且成本较高,而微生物转化法生产木糖醇具有条件温和、操作简便、副产物少、环境污染低等优点[2]。已知有多种假丝酵母均可以生产木糖醇,而热带假丝酵母是其中最佳的木糖醇生产菌,其利用木质纤维素水解物发酵生产木糖醇具有广阔的前景[3]。另一方面,热带假丝酵母也能利用有机废水生产单细胞蛋白[4,5]。该工艺不仅使有机废水循环利用,有益于环境保护,又能变废为宝,获得有价值的生物产品。目前热带假丝酵母已成功用于马铃薯淀粉废水[6]、红麻亚铵法制浆废液[7]、木薯淀粉酒精废液[8]、苎麻生物脱胶废水[9]等有机废水的生物处理以及用于分离脱臭馏出物中植物甾醇的研究[10]。在医学上,热带假丝酵母作为一种条件致病菌,其毒力与致病性对免疫功能不全患者的威胁仅次于白色念珠菌(Candida albicans)[11],欧洲将热带假丝酵母归类进致病微生物中,用于生产和研究需要相当高的安全标准[12]。
由于热带假丝酵母在工业生物技术、医学等领域具有重要的研究价值,利用代谢工程进行菌株改良受到了广泛关注。高效的遗传转化和基因删除技术是代谢工程育种的前提。近年来,国内外学者在热带假丝酵母的遗传转化和基因删除技术上开展了多方面的研究。Hara等[13]以潮霉素B抗性作为筛选标记,构建了热带假丝酵母URA3基因敲除突变盒,并对其等位基因的两个拷贝分别进行了破坏;他们还将该方法成功应用于敲除单个拷贝的POX4基因。高弘等[14]则分别利用潮霉素B抗性和G418抗性作为选择性标记构建了两个不同的基因敲除表达盒,转化热带假丝酵母,获得了两个拷贝的肉毒碱乙酰转移酶(Carnitine acetyltransferase, CAT)基因同时缺失的热带假丝酵母突变株。尽管如此,利用抗生素抗性作为筛选标记依仍然存在不少问题。首先,由于菌种、菌株的不同,其细胞壁结构、生理生化特性和遗传背景相差较大,抗性标记难以成为一种通用的筛选标记,在实际使用上有很大的限制[15]。其次,热带假丝酵母在密码子使用方面具有特殊性,常将CUG密码子(通常被翻译成亮氨酸)翻译成丝氨酸的特性[16],这就限制了外源性抗性基因作为选择性标记使用的效率。另外,在二倍体酵母的基因敲除中,往往需要多个抗性基因作为选择性标记来完成多拷贝靶基因的敲除,进一步限制了抗性标记的使用。
目前,热带假丝酵母的遗传转化主要利用尿嘧啶营养缺陷型作为选择性标记,并以此为基础完成了多个靶基因的敲除。Haas等[17]最早建立起以营养缺陷型菌株为基础的热带假丝酵母DNA整合系统。利用这套转化系统,Picataggio等[18]通过对POX4和POX5基因进行连续破坏,建立了在同一株热带假丝酵母细胞进行多基因连续敲除技术。然而该基因连续敲除系统中,每完成一次基因的单拷贝敲除,就需要通过突变或者分子手段使转化子重新回复成尿嘧啶缺陷型,操作十分繁琐。Alani等[19]在酿酒酵母中建立了一种利用在 URA3基因两段分别插入一段源于沙门氏菌(Salmonella)的hisG重复序列的方法,可快速重复利用选择标记。Ko等[20]将该方法应用于热带假丝酵母的基因敲除实验并获得了 XYL2基因单拷贝敲除的杂合子突变株和双拷贝敲除的纯合子突变株。
本文建立了基于尿嘧啶缺陷型标记基因重复利用的热带假丝酵母基因连续敲除方法,并成功敲除了二倍体细胞中的PDC基因。
1 材料和方法
1.1 菌种、质粒
热带假丝酵母ATCC20336和大肠杆菌(Escherichia coli)JM109由本实验室保存,质粒pCUB6由中科院上海生物化学研究所陈江野研究员馈赠。
1.2 尿嘧啶缺陷型突变株的筛选
[21],通过1-甲基-3-硝基-1-亚硝基胍(NTG)化学诱变法获得尿嘧啶缺陷型突变株。原始菌株C. tropicalis ATCC 20336在YPD培养基(酵母粉1%、蛋白胨2%、葡萄糖2%)中30℃,200 r/min摇瓶培养,细胞生物量OD600到1.0时,离心收集细胞并无菌去离子水洗涤一次。将细胞重悬在 10 mL YPD培养基中,倾入含1~4 mg NTG的无菌50 mL离心管中,37℃、200 r/min培养40 min。离心收集菌体,并用无菌去离子水洗涤2次后于YPD培养基中后培养3 h,然后用MM培养基(YNB 0.67%、硫酸铵1%、葡萄糖2%)洗涤2次,再转入MM培养基中饥饿培养6~8 h,加入制霉菌素使其终浓度达50 μg/mL。培养 2 h后,离心收集菌体,并用无菌去离子水洗涤2次,转入YPD培养基中培养10 h,取适量涂布FOA培养基(SM + 5-氟乳清酸0.1%)平板,30℃培养2 d。将长出的单菌落分别点种于MM和SM培养基(MM + 尿嘧啶0.006%)平板上,在MM培养基平板不长而在 SM培养基平板生长的菌株即初步确认为突变株,待进一步验证。
1.3 PDC基因敲除突变盒的构建
根据 NCBI中热带假丝酵母的 URA3基因(GenBank登录号:AB006207)设计上下游引物URA3-F和URA3-R(表1),以热带假丝酵母ATCC 20336菌株的基因组为模板,PCR扩增获得1.6 kb的URA3基因片段,插入pMD18-T载体中,获得重组质粒,命名为T-URA3。用限制性内切酶EcoRⅠ和PstⅠ消化质粒,回收URA3片段备用。根据NCBI中热带假丝酵母MYA-3404菌株的 PDC 基因(NCBI Reference Sequence: XM_002549483.1)设计引物 PDC-F1和PDC-R1(表1),以热带假丝酵母ATCC 20336菌株的基因组为模板,PCR获得1.7 kb的PDC基因片段,插入pMD18-T simple载体中,获得重组质粒,命名为Ts-PDC。然后以Ts-PDC为模板,PDCrFE和PDCrRP为引物,反向 PCR获得含有两段同源臂的PDC1-Ts-PDC1片段。用EcoRⅠ和PstⅠ消化后,与回收的URA3片段连接,获得重组质粒Ts-PDC1-URA3。
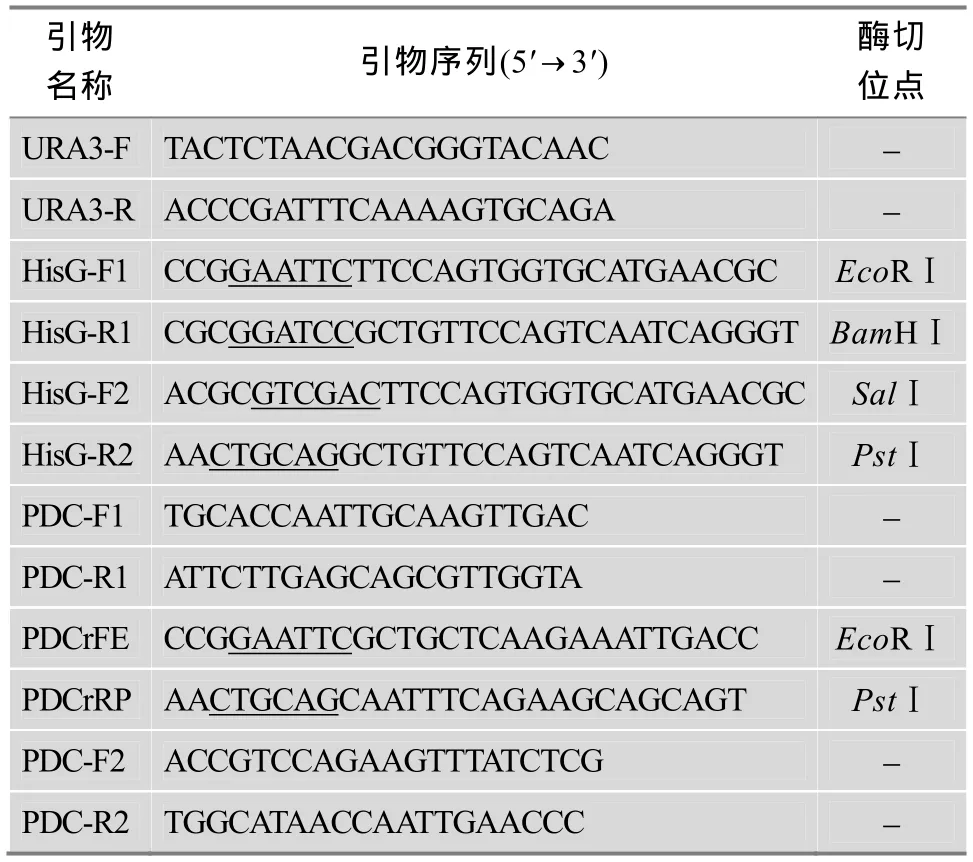
表1 PCR引物序列
根据文献[20]设计引物 HisG-F1和 HisG-R1、HisG-F2和HisG-R2两对引物(表1),以质粒pCUB6为模板,PCR获得大小均为1.1 kb的hisG1片段和hisG2片段,将hisG1插入Ts-PDC1-URA3上EcoRⅠ和BamHⅠ酶切位点之间,获得重组质粒。后将hisG2片段插入该重组质粒上的 SalⅠ和 PstⅠ酶切位点之间,获得带有 PDC1-hisG1-URA3-hisG2-PDC1片段(PHUHP片段)的重组质粒,命名为 Ts-PHUHP,重组质粒物理图谱如图1A所示。
根据扩增获得的 PDC基因的测序结果设计引物PDC-F2和PDC-R2,以热带假丝酵母ATCC 20336菌株的基因组为模板,PCR获得600 bp的PDCm片段。PDCm片段是PDC基因片段的中间部分,位于上述第一个敲除突变盒中用到的两段PDC同源臂片段之间。依上述方法,构建 Ts-PDCm-URA3,重组质粒物理图谱如图1B所示。本文所用的引物(表1)合成及测序由上海生工生物工程技术服务有限公司完成。分子克隆所用的工具酶均购自宝生物工程(大连)有限公司。
1.4 酵母的转化
氯化锂(LiCl)转化法参考文献[17]。将宿主菌培养至OD600为1.0左右,离心收集细胞,用TE溶液(10 mmol/L Tris-HCl,pH7.4、1 mmol/L EDTA)清洗一次,重悬在1 mL 100 mmol/L氯化锂中,30℃、200 r/min孵育1 h。取88 μL细胞,与10 μL DNA片段和2 μL鲑鱼精混匀,30℃孵育30 min。
再加入900 μL PEG3350溶液(40% PEG3350、100 mmol/L 氯化锂),30℃、200 r/min孵育1 h。然后42℃热激5 min,快速冷却至室温,离心收集细胞,无菌水清洗1次,涂布MM平板,30℃培养2~3 d。
醋酸锂(LiAc)转化法参考文献[20]。将宿主菌培养至OD600为1.0左右,离心收集细胞,用无菌去离子水清洗,重悬在1 mL醋酸锂溶液(0.1 mol/L醋酸锂、10 mmol/L Tris-HCl,pH7.6、1 mmol/L EDTA)中,取30 μL细胞,与50 μL DNA片段、5 μL鲑鱼精以及400 μL PEG8000溶液(50%的PEG8000溶于醋酸锂溶液中)混匀,30℃孵育30 min,42℃热激15 min,快速冷却至室温,离心收集细胞,用无菌去离子水清洗1次,涂布MM平板,30℃培养2~3 d。
1.5 PDC基因敲除与URA3基因的重复利用
热带假丝酵母细胞两个 PDC基因拷贝的敲除流程如图2所示。将转入PHUHP基因删除表达盒并成功整合于染色体上的热带假丝酵母转化子菌株接种于SM培养基中于30℃、200 r/min摇瓶培养,细胞生物量OD600达到1.0时,离心收集细胞,用无菌去离子水重悬后,涂布于2×FOA培养基(SM + 5-氟乳清酸0.2%)平板,于30℃培养2~3 d,将长出的单菌落转接于SM平板培养,提取染色体并PCR验证,将获得的 URA3标记基因剔除的突变株保藏并应用于第二轮转化。
1.6 PDC酶活测定
参考文献[22]制备细胞抽提物。酵母细胞接种于50 mL YPD或者SM液体培养基,于30℃、200 r/min培养16 h后,离心收集细胞。用10 mmol/L磷酸钠缓冲液(含2 mmol/L EDTA,pH7.5)洗2次,离心后用 3 mL 100 mmol/L磷酸钠缓冲液(含 2 mmol/L MgCl2,pH7.5)重悬。按工作2 s间歇冷却4 s的方式,超声破碎菌体,每15 min取样镜检观察细胞破碎情况。待观察到细胞破碎基本完全,12 000 r/min离心5 min,上清液即细胞抽提物。该上清液用于后续测定蛋白含量以及丙酮酸脱羧酶活性。
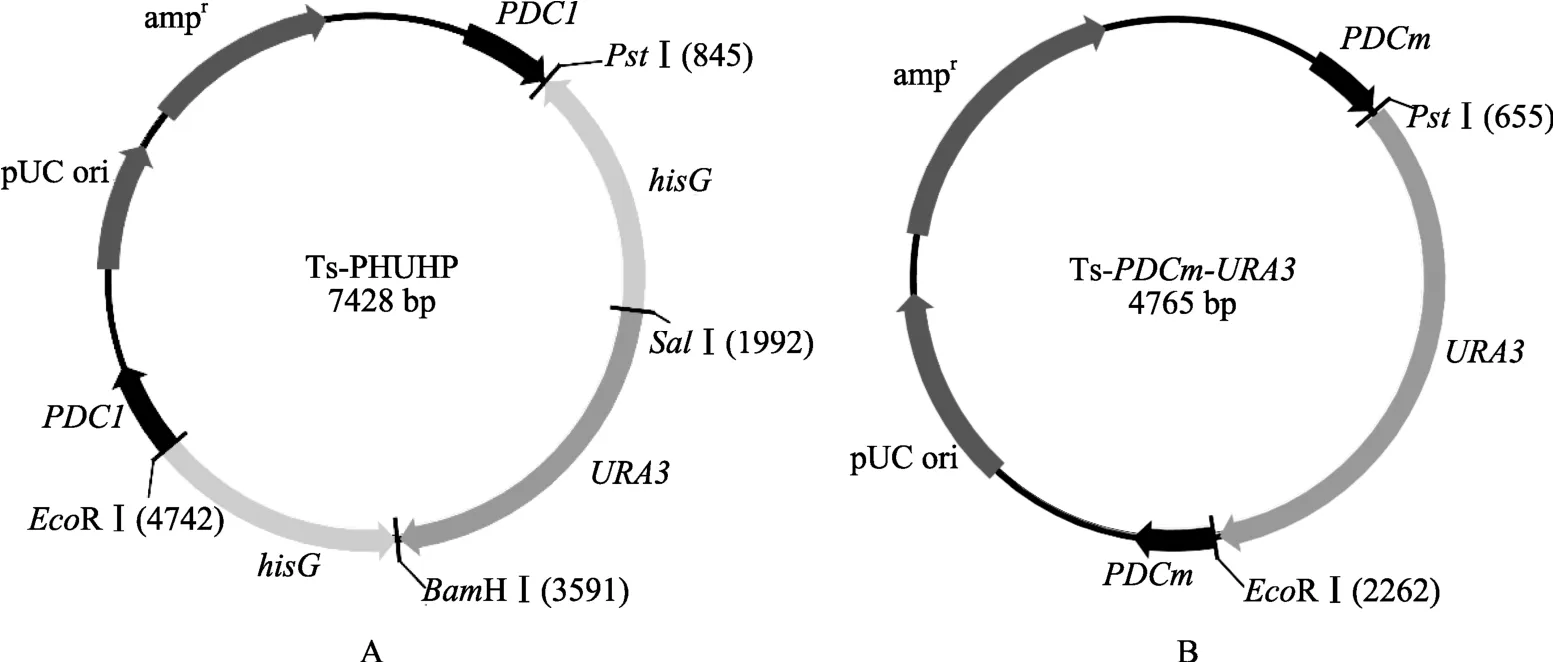
图1 重组质粒Ts-PHUHP和Ts-PDCm-URA3的物理图谱A:重组质粒Ts-PHUHP的物理图谱;B:重组质粒Ts-PDCm-URA3的物理图谱。
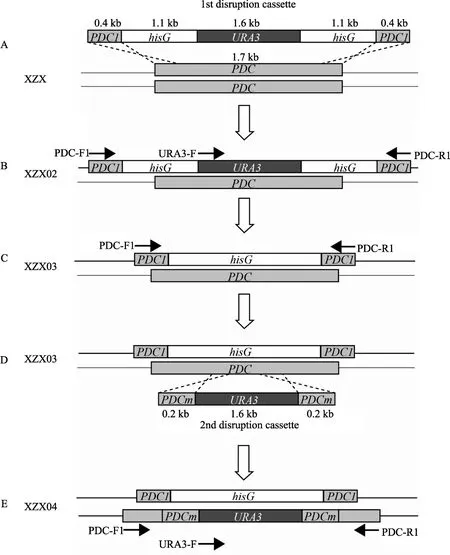
图2 PDC基因敲除流程图
按照文献[22]方法测定热带假丝酵母的PDC酶活。吸取2.7 mL柠檬酸缓冲液(200 mmol/L,pH6.0)、100 μL丙酮酸钠溶液(1 mol/L),50 μL β-NADH溶液(6.4 mmol/L)以及50 μL乙醇脱氢酶溶液(200 U/mL)于比色皿中混合均匀,作为实验组;吸取2.8 mL柠檬酸缓冲液(200 mmol/L,pH6.0)和100 μL丙酮酸钠溶液(1 mol/L)于比色皿中混合均匀作为空白对照组。两者经30℃保温5 min后,放入分光光度计,分别加入100 μL经适当稀释的细胞抽提物,迅速混合均匀,在波长340 nm处测定吸光值,每30 s记录一次数据,共测定3 min。计算每分钟吸光值的减少量,据此计算PDC比酶活。蛋白浓度用考马斯亮蓝染色法测定。
2 结果与分析
2.1 尿嘧啶缺陷型突变株的筛选
以热带假丝酵母ATCC 20336为出发菌株,经过11次诱变和FOA选择培养基筛选,先后共挑取127株单菌落进行营养缺陷型验证。将长出的菌落分别点种SM和MM培养基平板并进行突变株稳定性试验,最终鉴别出13株ura3/ura3突变株,选择其中3株,分别命名为C. tropicalis XZW、C. tropicalis XZX、C. tropicalis XZB。将原始菌株与3株营养缺陷型菌株分别划线于SM平板和MM培养基平板上,30℃培养4 d,生长状况如图3所示。突变株在MM培养基平板上不生长,而在 SM培养基平板上能够正常生长。
分别提取热带假丝酵母突变株 XZW、XZX、XZB的染色体DNA,PCR扩增获得突变株的URA3基因片段进行 DNA测序,并与来源于野生型菌株ATCC20336的URA3基因序列和蛋白质序列进行比较,结果见表 2。菌株 XZW 的氨基酸序列中,第203位的甘氨酸突变为天冬氨酸,第 261位的赖氨酸突变为精氨酸、第 265位的谷氨酰胺突变为亮氨酸;菌株XZX的氨基酸序列中,第8位的苏氨酸突变为丙氨酸,第25位的亮氨酸突变为异亮氨酸,第203位的甘氨酸突变为天冬氨酸;菌株XZB的氨基酸序列中,第 203位的甘氨酸突变为天冬氨酸、第261位的赖氨酸突变为精氨酸、第 265位的谷氨酰胺突变为亮氨酸。其共有的突变为第203位的甘氨酸突变为天冬氨酸,由此推测,第203位的甘氨酸可能是该酶的关键活性位点,突变会导致该酶功能失活。
2.2 PDC基因敲除突变盒的构建
分别构建重组质粒Ts-PHUHP(图1A)和Ts-PDCm-URA3(图1B)。重组质粒Ts-PHUHP经EcoRⅠ和PstⅠ双酶切作用后,凝胶电泳显示存在3.5 kb和3.9 kb的 DNA条带,分别与 PDC1-Ts-PDC1片段和 hisGURA3-hisG片段大小一致(图4A);利用EcoRⅠ和SalⅠ限制性内切酶作用于重组质粒,分别得到4.7 kb和2.7 kb的DNA条带,与预期的hisG-PDC1-Ts-PDC1和URA3-hisG片段大小一致(图4A)。结果表明基因敲除表达盒PHUHP构建成功。
利用EcoRⅠ和PstⅠ限制性内切酶双酶切重组质粒Ts-PDCm-URA3后,凝胶电泳显示释放出3.2 kb和1.6 kb的两个DNA片段,分别与预期的PDCm-Ts-PDCm片段和URA3基因大小一致(图4B)。表明成功构建了第二个基因敲除表达盒MUM。
2.3 热带假丝酵母PDC基因的敲除
PCR扩增基因敲除表达盒PHUHP(DNA结构示意图如图2A所示)并转化热带假丝酵母XZX宿主,涂布 MM培养基平板。将长出的单菌落转接培养,提取其染色体DNA作为模板进行PCR验证。用引物PDC-F1和PDC-R1扩增,获得4.7 kb的DNA片段条带和1.7 kb的DNA条带(图5A),测序发现这两个条带分别与PHUHP片段和PDC基因序列完全一致。而出发菌株C. tropicalis XZX的PCR结果仅有1.7 kb的PDC基因条带出现(图5A)。同时,用引物URA3-F和PDC-R1进行PCR扩增获得了3.1 kb的 DNA片段(图 5B),测序结果表明该片段与URA3-hisG- PDC1片段的序列完全一致。因此,可以确认基因敲除表达盒 PHUHP正确整合到染色体的PDC基因位点,其整合位点基因结构如图2B所示。该转化子命名为C. tropicalis XZX02。实验共挑取了19个单菌落进行PCR验证,其中14个成功整合在PDC基因位点,整合阳性率达74%(数据未列出)。
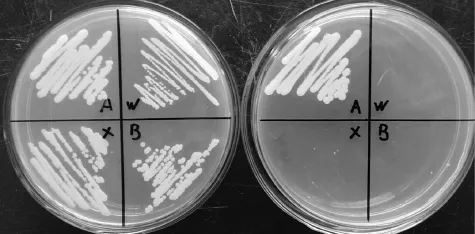
图3 原始株和突变株在MM(左)和SM(右)平板上的生长状况比较A: C. tropicalis ATCC20336; X: C. tropicalis XZX; W: C. tropicalis XZW; B: C. tropicalis XZB。
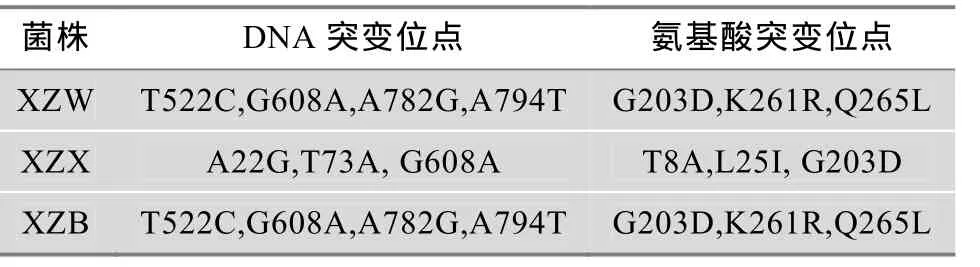
表2 热带假丝酵母营养缺陷菌株的URA3基因突变位点

图4 重组质粒的酶切鉴定A: Ts-PHUHP的酶切验证。1: Ts-PHUHP/EcoRⅠ+PstⅠ; 2: Ts-PHUHP/ EcoRⅠ+SalⅠ; M: DL 5000 DNA marker。B: Ts-PDCm-URA3的EcoRⅠ和PstⅠ双酶切验证。1: 酶切条带; M: DL 5000 DNA marker。
为获得 URA3标记基因从转化子 C. tropicalis XZX02染色体的 PDC基因位点丢失的突变株,将培养后的XZX02细胞涂布于2×FOA平板上,挑取单菌落进行PCR鉴定。用引物PDC-F1和PDC-R1进行PCR扩增分别获得2 kb的DNA片段和1.7 kb的DNA片段(图5A),经DNA测序分析表明其分别为PDC1-hisG-PDC1片段序列和PDC基因序列。进一步用引物URA3-F和PDC-R1进行PCR扩增,没有产物出现(图 5B)。推测原因可知,由于 PHUHP片段中的URA3基因和一个拷贝hisG序列在DNA复制过程中从染色体上丢失,引物对无法有效结合靶序列导致了该 PCR反应没有特异性产物出现。PCR鉴定结果确认筛选培养基上长出的单菌落细胞中插入于PDC基因位点的URA3标记基因已经丢失,丢失URA3基因后的突变株命名为C. tropicalis XZX03,染色体上PDC基因位点附近的遗传结构如图2C所示。
以菌株C. tropicalis XZX03为宿主,将第二个PDC基因删除表达盒MUM片段导入受体细胞,涂布MM培养基平板,挑取单菌落进行PCR鉴定。用引物PDC-F1和PDC-R1进行PCR扩增,分别获得2 kb的DNA片段和3 kb DNA片段(图5A),大小分别与PDC1-hisG-PDC1片段和包含PDCm-URA3-PDCm的DNA片段一致,DNA测序进一步验证了该推测的正确性。用引物URA3-F和PDC-R1进行PCR扩增,获得2.2 kb的DNA片段,DNA测序表明包含URA3-PDCm片段的序列。结果表明宿主的第二个PDC等位基因被 MUM 片段所替代,该突变株命名为 C. tropicalis XZX04,染色体上PDC基因结构如图2E所示。另外,评价了第二个PDC基因的敲除效率。从MM培养基平板分别挑取12株单菌落,经PCR及DNA测序验证其中6个单菌落细胞的PDC第二个等位基因被 MUM片段所取代,定点整合效率达50%(数据未列出)。
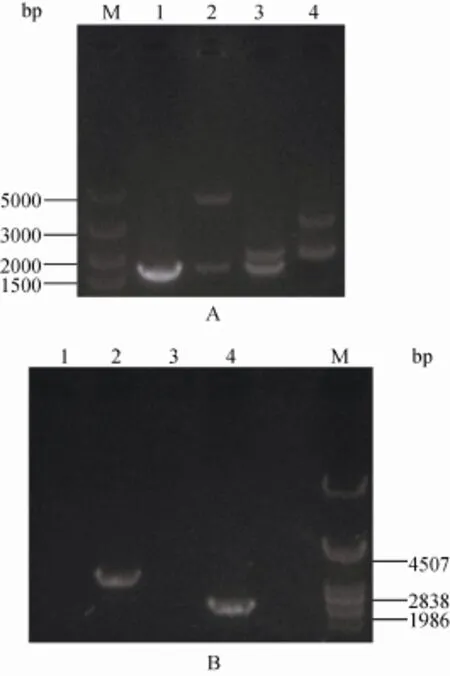
图 5 尿嘧啶缺陷株 XZX、突变株 XZX02、XZX03和XZX04的PCR验证A:以PDC-F1和PDC-R1为引物,各菌株染色体DNA为模板进行PCR验证。1: C. tropicalis XZX; 2: C. tropicalis XZX02; 3: C. tropicalis XZX03; 4: C. tropicalis XZX04; M: DL 5,000 DNA marker。B:以URA3-F和PDC-R1为引物,各菌株染色体DNA为模板进行 PCR验证。1: C. tropicalis XZX; 2: C. tropicalis XZX02; 3: C. tropicalis XZX03; 4: C. tropicalis XZX04; M: lambda DNA/PstI marker。
2.4 热带假丝酵母 C. tropicalis(pdc/pdc)突变株的表型鉴定
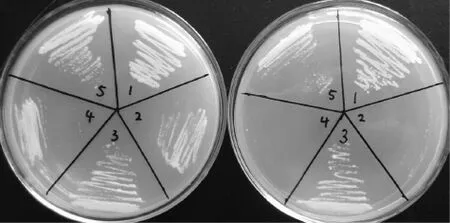
图6 原始菌株ATCC20336及衍生菌株在SM平板(左)和MM平板(右)上的生长状况比较1: C. tropicalis ATCC 20336; 2: C. tropicalis XZX; 3: C. tropicalis XZX02; 4: C. tropicalis XZX03; 5: C. tropicalis XZX04。
将原始菌株 ATCC 20336、尿嘧啶缺陷株 C. tropicalis XZX、突变株 C. tropicalis XZX02、C. tropicalis XZX03和C. tropicalis XZX04分别划线于SM平板和MM平板上,观察其生长状况,如图6所示。在 SM培养基平板上,所有热带假丝酵母菌株均能正常生长。当划线接种于MM培养基平板上时,仅有C. tropicalis ATCC 20336、转化子C. tropicalis XZX02和C. tropicalis XZX04具有相似的生长表型,而营养缺陷株C. tropicalis XZX和转化子C. tropicalis XZX03不能正常生长形成肉眼可见的菌落。生长表型实验进一步验证了不同转化子PDC基因位点的重组特征及二倍体细胞中两个PDC基因拷贝的敲除。
2.5 PDC基因缺失的功能鉴定
为了考察 PDC基因的缺失对突变株中酶活力的影响,分别测定了尿嘧啶缺陷型菌株C. tropicalis XZX、突变株 C. tropicalis XZX02、C. tropicalis XZX03和C. tropicalis XZX04的胞内PDC酶活力,结果如图7所示。C. tropicalis XZX的酶活最高,PDC基因单拷贝敲除菌株 C. tropicalis XZX02和 C. tropicalis XZX03的PDC比酶活相似,分别比出发菌株C. tropicalis XZX下降了20.56%和20.00%。PDC基因双拷贝敲除菌株 C. tropicalis XZX04的PDC比酶活相比出发菌株C. tropicalis XZX则下降了 43.60%。结果表明 PDC基因的敲除显著降低了胞内PDC酶活。但同时可以看到,即使PDC基因两个拷贝都敲除的 C. tropicalis XZX04胞内,仍有 56.30%的 PDC酶活。通过对热带假丝酵母基因组DNA序列分析发现,该酵母细胞中存在着至少3个编码PDC的同工酶基因。因此,我们推测突变株XZX04中 PDC同工酶提供了胞内相应的酶活并能在一定程度上功能互补缺失基因。
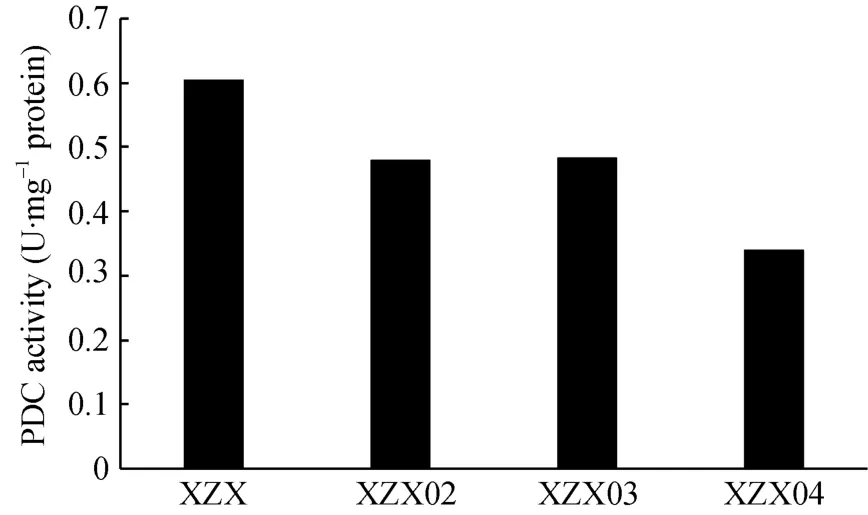
图7 热带假丝酵母转化子胞内PDC酶活分析XZX: C. tropicalis XZX; XZX02: C. tropicalis XZX02; XZX03: C. tropicalis XZX03; XZX04; C. tropicalis XZX04。
3 讨 论
本研究成功构建了一种适用于热带假丝酵母的基因敲除系统。热带假丝酵母是一种二倍体酵母,其每一个双拷贝基因的敲除,都需要构建两个不同的突变盒,以保证两个拷贝基因的彻底破坏,获得该基因缺失的纯合子突变株。如果每个突变盒都需要一个不同的标记基因进行多基因敲除时,寻找合适的标记基因会变得十分困难。因此,重复利用同一选择性标记进行多基因的删除是切实可行的策略。在染色体DNA复制过程中,两个近距离的重复片段会发生同源重组现象。在构建基因删除表达盒过程中,将来源于沙门氏菌的 hisG序列分别同向插入URA3基因的两侧,并转化宿主细胞。在DNA复制过程中,整合于宿主染色体上的hisG-URA3-hisG结构的DNA片段会发生hisG序列的重组从而剪切掉URA3基因和一个拷贝的hisG片段导致重组位点仅留下一个hisG片段[20]。利用这种同源重组原理,以“转化宿主菌、剔除URA3基因、更换同源臂再次转化宿主菌”的循环操作,能够实现对酵母的多个靶基因的连续敲除。
本研究重复利用 URA3基因作为选择标记,成功敲除了热带假丝酵母二倍体细胞的PDC基因,并对该基因功能进行了初步验证。PDC基因可在无氧条件将丙酮酸转化为乙醛,并在乙醛脱氢酶的作用下进一步代谢为酒精,是热带假丝酵母碳代谢途径上的关键酶。另外,PDC基因的存在,可能还与假丝酵母的致病性有一定关联[23]。作为对热带假丝酵母 URA3基因重复利用可行性的初步探索,本研究仅对一个基因的两个拷贝进行敲除,故第二个敲除突变盒中并未插入hisG重复序列,没有对URA3基因进行再次剔除。在实际应用中,如需对多个不同基因进行敲除,可在每一个敲除突变盒中均插入hisG重复序列,通过两次筛选消除URA3基因,即可实现 URA3标记的多次重复利用。另外,本文还比较了醋酸锂转化法与氯化锂转化法的转化效率。结果表明氯化锂转化法的转化效率较高(数据未列出)。
利用本研究建立的基因敲除系统能够高效地敲除热带假丝酵母的目的基因,为菌株的代谢工程研究提供了技术手段,也可以作为其他酵母遗传转化系统的借鉴。
参考文献
[1] 陈远童. 十二碳二元酸工业生产试验研究. 微生物学通报, 1998, 25(4): 244-244.
[2] 焦静雨, 吴绵斌, 赵炯烽, 林建平, 杨立荣. 基因工程技术改造木糖醇生产菌株的研究进展. 中国生物工程杂志, 2012, 32(11): 124-131.
[3] Sánchez S, Bravo V, García JF, Cruz N, Cuevas M. Fermentation of D-glucose and D-xylose mixtures by Candida tropicalis NBRC 0618 for xylitol production. World J Microbiol Biotechnol, 2008, 24(5): 709-716.
[4] Adedayo MR, Ajiboye EA, Akintunde JK, Odaibo A. Single cell proteins: as nutritional enhancer. Adv Appl Sci Res, 2011, 2(5): 396-409.
[5] Gao YR, Li DP, Liu Y. Production of single cell protein from soy molasses using Candida tropicalis. Ann Microbiol, 2012, 62(3): 1165-1172.
[6] 张玉斌, 王友玲. 热带假丝酵母菌处理马铃薯淀粉废水的研究. 安徽农业科学, 2013, 41(20): 8698-8699, 8702.
[7] 甄娜, 何连芳, 周景辉. 热带假丝酵母处理红麻亚铵法制浆废液的研究. 造纸科学与技术, 2007, 26(2): 26-29, 33.
[8] 刘继栋, 付灿, 王同阳. 热带假丝酵母在木薯淀粉酒精废液治理中的应用研究. 中国酿造, 2009(7): 121-123.
[9] 王军, 郝新艳, 殷朝敏, 曾庆福. 不同氮源对热带假丝酵母处理苎麻生物脱胶废水的影响. 工业水处理, 2013, 33(3): 29-32.
[10] Fernandes P, Cabral J M S. Phytosterols: applications and recovery methods. Bioresour Technol, 2007, 98(12): 2335-2350.
[11] Gleeson MA, Haas LO, Cregg JM. Isolation of Candida tropicalis auxotrophic mutants. Appl Environ Microb, 1990, 56(8): 2562-2564.
[12] Huf S, Krügener S, Hirth T, Rupp S, Zibek S. Biotechnological synthesis of long-chain dicarboxylic acids as building blocks for polymers. Eur J Lipid Sci Tech, 2011, 113(5): 548-561.
[13] Hara A, Arie M, Kanai T, Matsui T, Matsuda H, Furuhashi K, Ueda M, Tanaka A. Novel and convenient methods for Candida tropicalis gene disruption using a mutated hygromycin B resistance gene. Arch Microbiol, 2001, 176(5): 364-369.
[14] 高弘. 生物催化生产十三碳二元酸中β-氧化途径的代谢调控[学位论文]. 北京: 清华大学, 2005.
[15] 龚毅, 蒋华, 杨胜利. 新的热带假丝酵母载体-宿主系统的建立. 生物工程学报, 1997, 13(3): 309-312.
[16] Ueda T, Suzuki T, Tokogawa T, Nishikawa K, Watanabe K. Unique structure of new serine tRNAs responsible for decoding leucine codon CUG in various Candida species and their putative ancestral tRNA genes. Biochimie, 1994, 76(12): 1217-1222.
[17] Haas LO, Cregg JM, Gleeson MA. Development of an integrative DNA transformation system for the yeast Candida tropicalis. J Bacteriol, 1990, 172(8): 4571-4577.
[18] Picataggio S, Deanda K, Mielenz J. Determination of Candida tropicalis acyl coenzyme A oxidase isozyme function by sequential gene disruption. Mol Cell Biol, 1991, 11(9): 4333-4339.
[19] Alani E, Cao L, Kleckner N. A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains. Genetics, 1987, 116(4): 541-545.
[20] Ko BS, Kim J, Kim JH. Production of xylitol from D-xylose by a xylitol dehydrogenase gene-disrupted mutant of Candida tropicalis. Appl Environ Microb, 2006, 72(6): 4207-4213.
[21] 陈珺, 林海, 王正祥, 诸葛健. 双重筛选产甘油假丝酵母营养缺陷型菌株及其发酵性能. 无锡轻工大学学报, 2000, 19(2): 95-99.
[22] 高年发, 邓旭衡, 王德培, 李磊. 酿酒酵母丙酮酸脱羧酶活性测定的研究. 中国酿造, 2011(3): 128-130.
[23] Otzen C, Fleck CB, Brock M. Role of the glyoxylate cycle in pathogenesis of the yeast Candida albicans and the filamentous fungus Aspergillus fumigatus: Pyruvate decarboxylase activity provides the key for metabolite flow. Mycoses, 2012, 55(4): 17-18.
(责任编委: 吕 红)
Development of a genetic transformation system for Candida tropicalis based on a reusable selection marker of URA3 gene
Zheng Xiang1, Xianzhong Chen1, Lihua Zhang1, Wei Shen1, You Fan1, Maolin Lu2
1. Key Lab of Industrial Biotechnology, Education Ministry, School of Biotechnology, Jiangnan University, Wuxi 214122, China;
2. Jiangsu Institute of Microbiology Co. Ltd., Wuxi 214063, China
Candida tropicalis, a diploid asporogenic yeast, is frequently utilized in industrial applications andresearch studies. However, the low efficiency of genetic transformation limits the strain improvement by metabolic engineering. A reliable transformation and efficient deletion of target gene are prerequisite for molecular improvement of C. tropicalis. In this study, an efficient approach for genetic transformation of C. tropicalis was developed based on the URA3 gene as a reusable selection marker and both of PDC allele genes encoding pyruvate decarboxylase were successfully deleted by this approach. Firstly, an auxotrophic mutant strain of C. tropicalis XZX which is defective in orotidine-5′-phosphate decarboxylase (URA3) was isolated by chemical mutagenesis combined with nystatin enrichment selection and 5-fluoro-orotic acid (5-FOA) resistance selection using C. tropicalis ATCC 20336 as the parent strain. Then, the first PDC deletion cassette PDC1-hisG-URA3-hisG- PDC1 (PHUHP) which contains a 1.6 kb URA3 marker gene, two copies of 1.1 kb Salmonella hisG fragments and homologous arms of target gene was constructed and transformed into C. tropicalis XZX cells. Transformants with a single copy of PDC deleted were isolated and identified by PCR and DNA sequencing, which was designated as C.tropicalis XZX02. The C.tropicalis XZX02 cells were spread on the minimal medium containing 5-FOA to generate mutant C. tropicalis XZX03 in which URA3 marker gene was excised from PHUHP fragment integrated into the PDC gene site. The second PDC gene deletion cassette PDCm-URA3-PDCm (MUM) was constructed and transformed into C. tropicalis XZX03 to generate C.tropicalis XZX04 in which both of PDC allele genes were deleted. All strains were confirmed by PCR and DNA sequencing. This efficient genetic transformation approach laid a foundation for further metabolic engineering of C. tropicalis.
Candida tropicalis; genetic transformation; homologous recombination; URA3 gene; gene disruption
2014-04-11;
2014-06-10
江苏省科技支撑计划项目(编号:BE2012618)资助
项峥,硕士研究生,专业方向:微生物遗传育种。E-mail: wq5513031@126.com
陈献忠,博士,副教授,研究方向:微生物遗传育种与代谢工程。E-mail: xzchen@jiangnan.edu.cn
10.3724/SP.J.1005.2014.1053
时间: 2014-9-24 10:35:09
URL: http://www.cnki.net/kcms/detail/11.1913.R.20140924.1035.003.html
