Co、Ni-锯齿型石墨烯纳米带体系的磁性与电子结构研究
2014-05-25韦嘉佳郑旭明
韦嘉佳,吴 韬,郑旭明
(1.浙江理工大学理学院,杭州310018;2.浙江大学理学院,杭州310027)
Co、Ni-锯齿型石墨烯纳米带体系的磁性与电子结构研究
韦嘉佳1,吴 韬2,郑旭明1
(1.浙江理工大学理学院,杭州310018;2.浙江大学理学院,杭州310027)
采用自旋极化的密度泛函理论计算方法研究Co、Ni吸附的锯齿型石墨烯纳米带(n=6,8,10)的几何构型、电子结构与磁性。在真空环境里,吸附过渡金属面后,锯齿型石墨烯纳米带发生弯曲,金属面大多不平整,且在n =8,10的锯齿型石墨烯纳米带上Co、Ni原子倾向于团聚成立体团簇。所有的Co、Ni-锯齿型石墨烯纳米带体系都是金属性的。计算结果表明,石墨烯纳米带的类一维性和边界形状将影响多个金属原子吸附后的堆积构型,同时锯齿型石墨烯纳米带作为吸附底物使吸附的过渡金属产生与无基底支持的二维、三维金属体系和石墨基底上吸附的过渡金属不同的磁性。
锯齿型石墨烯纳米带;3d过渡金属;密度泛函理论;磁性;电子结构;VASP
0 引 言
按照不同的方向切割石墨烯将得到边界形状不一样的石墨烯纳米带,石墨烯纳米带分为扶手椅和锯齿型两种[1]。扶手椅型石墨烯纳米带(armchair graphene nanoribbons,AGNRs)是非磁性的半导体,锯齿型石墨烯纳米带(zigzag graphene nanoribbons,ZGNRs)有反铁磁性和铁磁性的两种状态。反铁磁性的锯齿型石墨烯纳米带是基态,为半导体,它的两个边界之间是反铁磁耦合的。铁磁性的锯齿型石墨烯纳米带是激发态,为金属,它的两个边界之间是铁磁耦合的。石墨烯纳米带的电子结构可通过吸附过渡金属(transition metal,TM)改变,因此过渡金属修饰的石墨烯纳米带吸引了很多的研究兴趣[2-3]。
碳基底上吸附的过渡金属原子层的丰富磁性有望满足未来的自旋电子器件对材料的要求。Krüger等[4]研究比较了三维碳基底石墨吸附单层3d过渡金属前后的磁性差异,发现吸附后过渡金属的磁矩大幅减小,Mn吸附后基态由反铁磁性变为了亚铁磁性,而无基底支持的和石墨上吸附的Fe、 Co、Ni金属单层都是铁磁性的。Zanella等[5]对二维石墨烯碳基底上吸附的Ti金属单层和Fe金属单层进行了研究,发现Ti金属单层是非磁性的,Fe金属单层是铁磁性的,在石墨烯表面上Ti倾向于均匀分布而Fe倾向于团聚。覆盖于石墨烯上的非磁性Pd金属单层被Uchoa等[6]发现当存在一定电压U时,能产生不稳定的铁磁性,这种铁磁性是金属-碳杂化作用造成的。更复杂的过渡金属-碳体系也被讨论了,Zhou等[7]研究了3d过渡金属插入的双层石墨烯三明治体系的几何构型、电子结构和磁性,发现石墨烯双层倾向于AB堆叠模式而非AA堆叠模式,Fe、Co、Ni原子倾向吸附在C-C键中心,而V原子倾向吸附于碳原子正上方;Fe和Co的AB堆叠的三明治体系是铁磁性的金属,而Ni的AB堆叠的三明治体系是非磁性的半导体,V的AB堆叠的三明治体系则是非磁性的金属。
然而,还没有研究对类一维石墨烯纳米带上吸附的过渡金属原子层的构型和磁性进行讨论,为了探究尺度效应对碳基底上吸附的多个金属原子的磁性的影响,本论文将对Co、Ni-锯齿型石墨烯纳米带体系的结构、磁性与电子结构进行研究和讨论。
1 计算方法
Co、Ni锯齿型石墨烯纳米带体系(Co、Ni-ZGNR)的电子结构由采用投影缀加平面波(projector augmented-wave,PAW)的第一性原理的自旋极化密度泛函理论方法的VASP程序包计算获得。电子交换和关联势选用广义梯度近似(generalized gradient approximation,GGA)[8]的Perdew-Burke-Ernzerhof(PBE)[9]泛函进行近似计算获得。所有的计算都在4.92×30×15Å的超晶胞里进行,每个超晶胞包含2个重复单元的晶胞,内有氢钝化的锯齿型石墨烯纳米带,石墨烯纳米带上方覆盖3 d过渡金属层。对于原子的初始位置,每个过渡金属原子被放置在结构优化后的纯净石墨烯纳米带的每一个碳六元环中心上方,过渡金属单原子层与石墨烯纳米带平面之间的距离约为1.53Å。每个超晶胞中含有4个参与钝化的氢原子,含有宽度为n=6,8,10的石墨烯纳米带(Zig6,Zig8,Zig10)的超晶胞里对应分别含有4,6,8个过渡金属原子,因此对应的TM/C原子个数比例分别为0.333、0.375、0.400。截断能设为550.00 eV,原子受力的收敛值为0.02 eV/Å。沿着石墨烯纳米带生长方向的布里渊区Monkhorst-Pack的K点网格为9×1×1。每个过渡金属的结合能用以下公式计算:Eb=(EZGNR+nETM-Etoal)/n,其中EZGNR是结构优化后纯净石墨烯纳米带的能量,n是体系中包含的金属原子个数,ETM是单个独立过渡金属原子在15×15×15Å的立方真空盒子里时的能量,Etotal是吸附后的金属原子与石墨烯纳米带的总能量。金属原子的初始磁性猜测设置为铁磁性和反铁磁性两种,记录每种设置的收敛体系总能量和磁性,并比较它们的能量。体系的激发态比基态有更高的能量值,或者由于不稳定而无法收敛得到激发态。一些体系不会收敛到铁磁性或反铁磁性,而是亚铁磁性。
2 结果与讨论
优化后的Co、Ni-ZGNR的几何构型的俯视图和侧视图如图1所示。图1表明,纳米带因吸附多个过渡金属原子而发生了弯曲,并在n=8,10的Co、Ni-ZGNR体系内都产生了四面体金属立体团簇结构,说明锯齿型石墨烯纳米带的磁性和锯齿型的边界形状诱导Co、Ni立体团簇产生。表1列出了Co、Ni-ZGNR基态的电传导性与磁性。表2列出了Co、Ni-ZGNR中每个金属原子的磁矩和超晶胞的总磁矩,表2中编号表示金属原子的初始吸附位点,如图2所示。锯齿型石墨烯纳米带吸附过渡金属后,锯齿型石墨烯纳米带的磁性和C、H原子的磁矩受到金属原子的影响,与纯净的锯齿型石墨烯纳米带情况相异,过渡金属-锯齿型石墨烯纳米带体系(TM-ZGNR)的磁矩主要来自于过渡金属原子的贡献,C、H原子的磁矩远小于过渡金属原子的磁矩。图3是Co、Ni-ZGNR的分波态密度,虚线和实线分别表示石墨烯纳米带和过渡金属原子的分波态密度。由于过渡金属和石墨烯纳米带的强烈杂化,所有的Co、Ni-ZGNR的电传导性都是金属性的,杂化作用主要是由TM-d轨道和C-pz(π)轨道的互相作用引起的。
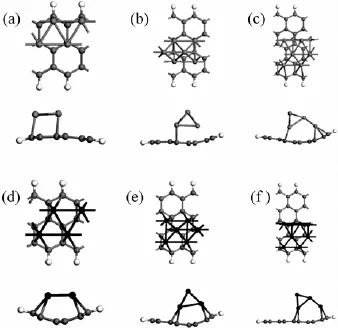
图1 优化的TM-ZGNR构型
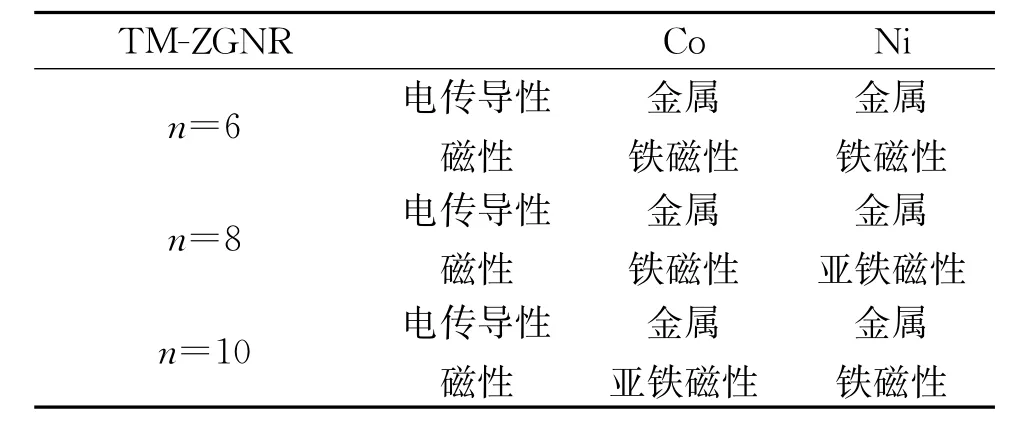
表1 TM-ZGNR基态的电传导性与磁性
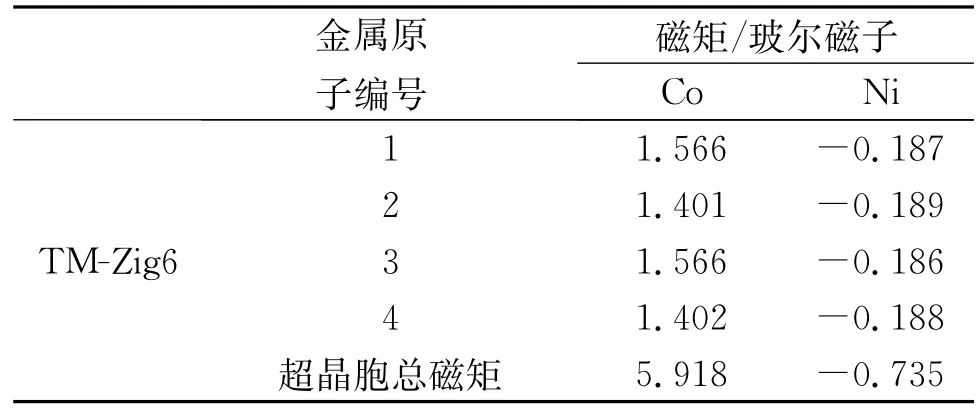
表2 TM-ZGNR中金属原子的磁矩与超晶胞总磁矩
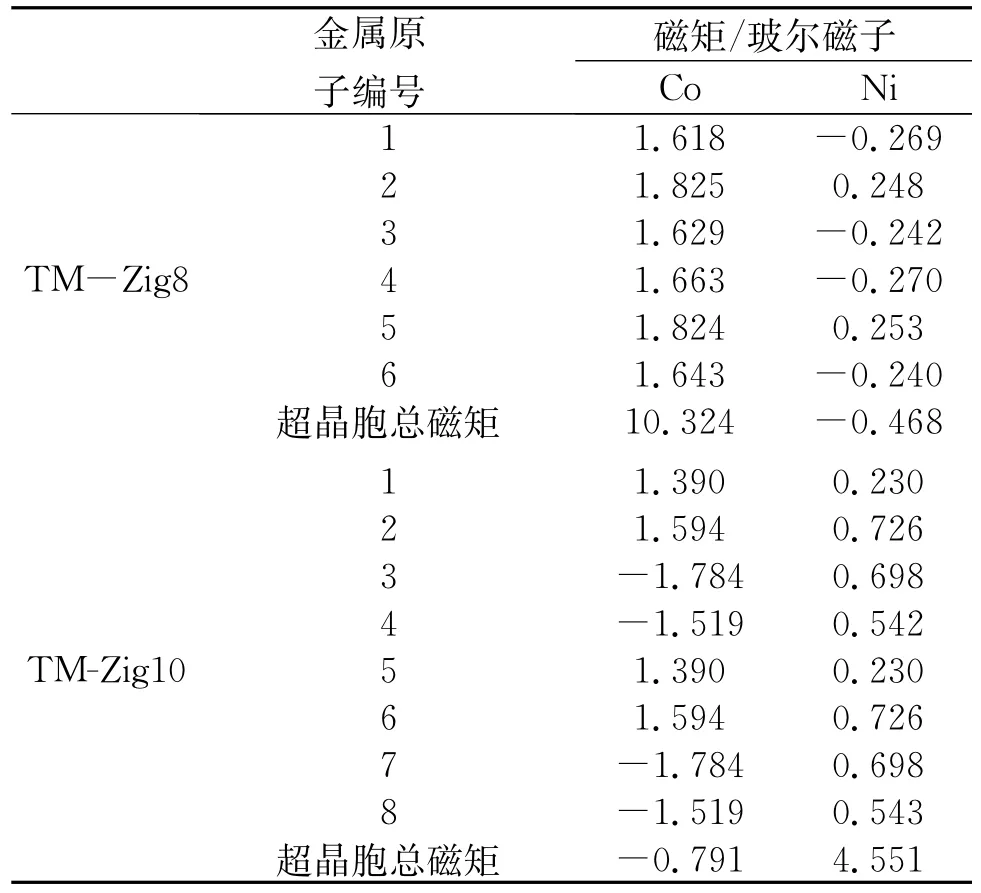
表1续
图2 过渡金属初始吸附位置编号
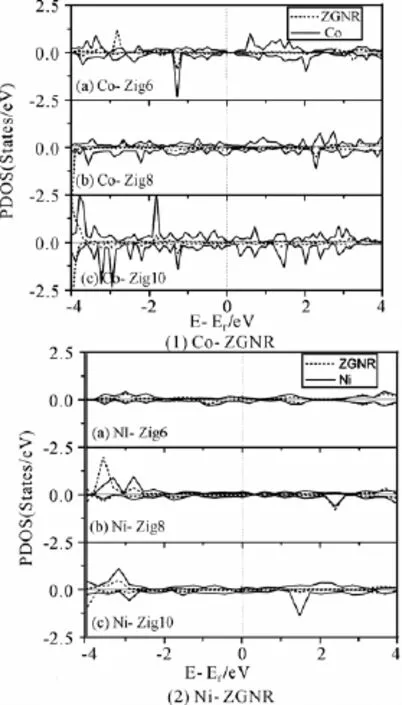
图3 Co,Ni-ZGNR的分波态密度
Co在体心立方晶系[10]和独立的六角晶格金属单原子层里都是铁磁性的,在石墨(0001)晶面上吸附的六角晶格Co金属单原子层也是铁磁性的[11]。Co-Zig6和Co-Zig8是铁磁性的,而Co-Zig10是亚铁磁性的。弯曲的类一维锯齿型纳米带作为吸附底物改变了Co原子层的几何结构,从而影响了Co原子的磁性,使其体现出与三维和二维Co体系不同的磁性。尽管吸附的Co原子层是铁磁性的,但Co-Zig6内的锯齿型石墨烯纳米带的边界仍有一定程度的反铁磁耦合,说明Co原子层把半导性的反铁磁锯齿型石墨烯纳米带的电传导性调控成金属性的了。而Co-Zig8和Co-Zig10内的锯齿型石墨烯纳米带的边界既非反铁磁耦合也非铁磁耦合,说明纳米带本身的磁性受到金属层的影响较大,失去了纯净锯齿型石墨烯纳米带原本的磁耦合性质。Co-Zig6里的Co金属层是光滑的,石墨烯纳米带背离Co原子弯曲,体系内每个Co原子都吸附在碳原子的正上方。Co-Zig8和Co-Zig10的Co金属层是不平整的,且有四面体立体团簇结构,石墨烯纳米带朝着Co原子弯曲。Co-Zig8和Co-Zig10内有类似体心立方晶体的原子排序情况,Co-Co键形成四元环,另有Co原子吸附在四元环中心的正上方。Co-ZGNR内C-Co键约为2.0~2.3Å,而Co-Co键约为2.3~2.5Å。对于Co-Zig6和Co-Zig8体系,都只存在铁磁性的基态。对于Co-Zig10体系,存在铁磁性的激发态,能量比亚铁磁性的基态高1.568 eV。
Ni在体心立方晶系[10]和独立的六角晶格金属单原子层里都是铁磁性的,在石墨(0001)晶面上吸附的六角晶格Ni金属单原子层也是铁磁性的[11]。Ni-Zig6和Ni-Zig10是铁磁性的,而Ni-Zig8是亚铁磁性的。弯曲的一维锯齿型纳米带作为吸附底物也改变了Ni原子层的几何结构,从而影响Ni原子的磁性。铁磁性的Ni-Zig6内的锯齿型石墨烯纳米带的边界有较弱的铁磁耦合,H原子没有磁矩,C原子的磁矩比纯净的锯齿型石墨烯纳米带的情况小。而Ni-Zig8和Ni-Zig10内的锯齿型石墨烯纳米带的边界既非反铁磁耦合也非铁磁耦合,说明纳米带本身的磁性受到金属层的影响较大,失去了纯净锯齿型石墨烯纳米带原本的磁耦合性质。三个Ni-ZGNR体系里石墨烯纳米带都朝着Ni原子弯曲。Ni-Zig6里的Ni单金属层是光滑的,Ni原子磁矩绝对值的平均值要低于Ni-Zig8和Ni-Zig10体系的情况。与Co-ZGNR的情况类似,Ni-Zig8和Ni-Zig10内也有类似体心立方晶体的原子排序情况。Ni-ZGNR内C-Ni键约为2.0~2.2Å,而Ni-Ni键约为2.3~2.5Å。对于Ni-Zig6,存在反铁磁性的激发态,能量只比铁磁性的基态高0.007 5 eV,在该激发态中Ni原子的磁矩非常小,绝对值为0.011μB,磁矩几乎消失。对于Ni-Zig8体系,只存在亚铁磁性的基态。对于Ni-Zig10体系,只存在铁磁性的基态。
图4显示了Co、Ni-ZGNR体系里单个过渡金属的结合能。Co在Zig8和Zig10上的吸附能比Zig6上的大,在Zig8和Zig10上的吸附能相差不多;Ni在石墨烯纳米带上的吸附能随纳米带宽度增加递增。宽度增加促进了过渡金属与石墨烯纳米带的杂化作用,更大的π电子离域范围更有利于过渡金属的3 d电子向石墨烯纳米带转移,同时金属团簇的生成降低了TM-ZGNR的总能,也使结合能增加。Ni-ZGNR体系的单原子吸附能比Co-ZGNR的大,这是因为Ni与ZGNR的杂化程度比Co与ZGNR的杂化程度高,从图3里的分波态密度可以看出Ni与石墨烯纳米带的态密度峰重合程度比Co的情况高。
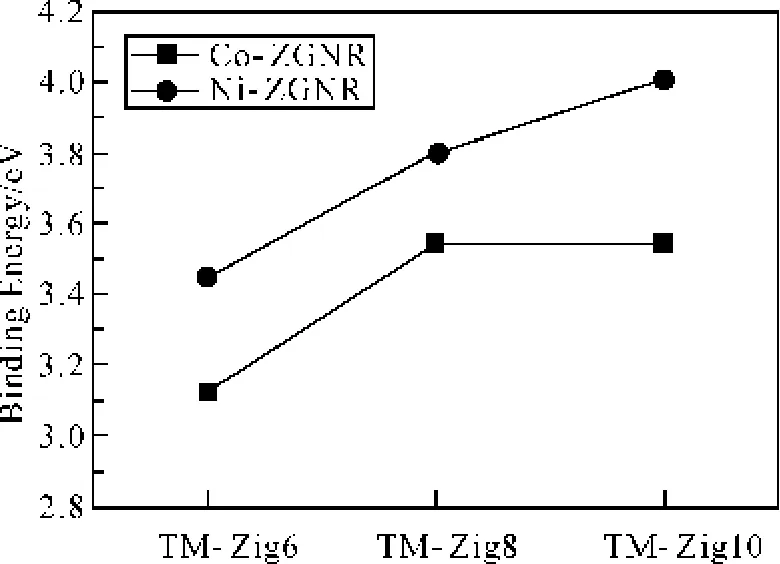
图4纳 米带上吸附的单个过渡金属结合能
3 结 论
采用自旋极化的密度泛函理论计算方法研究了Co、Ni吸附的锯齿型石墨烯纳米带体系(n=6,8,10)的几何构型、电子结构与磁性:
a)吸附多个过渡金属原子使锯齿型石墨烯纳米带结构发生弯曲,在n=8,10的Co、Ni-ZGNR里有四面体金属立体团簇结构产生,多个金属原子在石墨烯纳米带上的堆积结构受到石墨烯纳米带的类一维性和边界形状的影响;
b)所有的Co、Ni-ZGNR体系都呈现金属性;
c)锯齿型石墨烯纳米带上过渡金属独特的堆积构型使其产生与无基底支持的二维、三维金属体系以及石墨基底上吸附的过渡金属不同的磁性。
[1]Young W S,Marvin L C,Steven G L.Energy gaps in graphene nanoribbons[J].Phys Rev Lett,2006,97(21):216803:1-4.
[2]Longo R C,Carrete J,Gallego L J.Ab initio study of 3d,4d,and 5d transition metal adatoms and dimers adsorbed on hydrogen-passivated zigzag graphene nanoribbons[J].Phys Rev B,2011,83(23):1-9.
[3]Kan E J,Xiang H J,Yang J L,et al.Electronic structure of atomic Ti chains on semiconducting graphene nanoribbons:a first-principles study[J].J Chem Phys,2007,127(16):1-5.
[4]Krüger P,Rakotomahevitra A,Parlebas J C,et al. Magnetism of epitaxial 3d-transition-metal monolayers on graphite[J].Phys Rev B,1998,57(9):5276-5280.
[5]Zanella I,Fagan S B,Mota R,et al.Electronic and magnetic properties of Ti and Fe on graphene[J].J Phys Chem C,2008,112(25):9163-9167.
[6]Uchoa B,Lin C Y,Neto A C.Tailoring graphene with metals on top[J].Phys Rev B,2008,77(3):1-5.
[7]Zhou J,Wang L,Qin R,et al.Structure and electronic and transport properties of transition metal intercalated graphene and graphene-hexagonal-Boron-Nitride bilayer[J].J Phys Chem C,2011,115(51):25273-25280.
[8]Perdew J P,Chevary J A,Vosko S H,et al.Atoms,molecules,solids,and surfaces:applications of the generalized gradient approximation for exchange and correlation[J].Phys Rev B,1992,46(11):6671-6687.
[9]Perdew J P,Burke K,Ernzerhof M.Generalized gradient approximation made simple[J].Phys Rev Lett,1996,77(18):3865-3868.
[10]Moruzzi V,Marcus P.Magnetism in bcc 3d transition metals:onset and approach to the Hund’s-rule limit[J].Phys Rev B,1998,38(3):1613-1620.
[11]Kruger P,Rakotomahevitra A,Parlebas J C,et al. Magnetism of epitaxial 3d-transition-metal monolayers on graphite[J].Phys Rev B,1998,57(9):5276-5280.
Research on Magnetism and Electronic Structure of Co and Ni-Zigzag Graphene Nanoribbon System
WEI Jia-jia1,WU Tao2,ZHENG Xu-ming1
(1.School of Sciences,Zhejiang Sci-Tech University,Hangzhou 310018,China;2.School of Sciences,Zhejiang University,Hangzhou 310027,China)
This paper studies the geometrical configuration,electronic structure and magnetism of Co and Ni-zigzag graphene nanoribbons(n=6,8,10)with density functional theory computing method of spin polarization.In vacuum environment,after adsorbing transition metal surface,zigzag graphene nanoribbons bend.Most metal surfaces are not smooth and Co and Ni atoms on zigzag graphene nanoribbons(n= 8 and 10)tend to form aggregate cluster.All Co and Ni-zigzag graphene nanoribbon systems are metallic. The computing result shows that single-dimension property and edge contour of graphene nanoribbons will influence the stack configuration after the adsorption of multiple metal atoms.Meanwhile,as adsorption substrate,zigzag graphene nanoribbons make adsorbed transition metal produce magnetism different from two-dimensional and three-dimensional metal systems without substrate support and transition metal adsorbed on graphite substrate.
zigzag graphene nanoribbons;3d transition metal;density functional theory;magnetism;electronic structure;VASP
O641.121
A
(责任编辑:许惠儿)
1673-3851(2014)01-0098-04
2013-03-08
韦嘉佳(1986-),女,广西百色人,硕士研究生,主要从事石墨烯相关体系的第一性原理计算研究。
