碳水化合物活性酶数据库(CAZy)及其研究趋势
2014-05-04陈冠军张怀强王禄山
王 帅,陈冠军,张怀强,王禄山
(山东大学 微生物技术国家重点实验室,济南 250100)
碳水化合物亦称糖类化合物,是自然界存在最多、分布最广的一类重要有机化合物,是一切生物体维持生命活动所需能量的主要来源。在自然界中,糖类分子构型多样,糖分子之间化学键有多种类型,几乎所有生物大分子(糖分子、蛋白质、脂质和核酸等)都可以被糖基化,因此,作用于各种糖复合物、寡糖和多糖等碳水化合物的酶类就构成了地球上结构最多样的蛋白质集合,亦被定义为碳水化合物酶簇[1]。
碳水化合物酶类按功能分类主要包括糖苷水解酶类(EC 3.2.1.-)、糖基转移酶类(EC 2.4.-.-)、多糖裂解酶类(EC 4.2.2.-)等,这种系统命名原则及系统编号由国际酶学委员会(EC)制定,由其编号就可给出该酶分子类型及其催化反应性质,这是酶学研究的基础。一种酶分子一般只有一个名称及一个EC编号。然而最新研究表明,一种酶分子常常可催化一种以及以上类型的反应,即酶分子具有多功能性(promiscuity)或非特异性[2],特别是作用于复杂多糖的糖苷水解酶类,它们的底物专一性常常都不高。例如Bacillus licheniformis ATCC 14580分泌的1种内切-β-1,4-葡聚糖酶(GenBank登录号AAU42138.1),它既具有内切纤维素酶活性(EC 3.2.1.4),也 具 有 木 葡 聚 糖 酶 活 性 (EC 3.2.1.151)[3]。酶分子底物专一性不高,功能分析时就需要利用多种底物进行功能的测定,这就给酶分子功能的研究带来极大的工作量[4]。
新一代高通量测序技术的发展使得测序成本急剧降低,产生了可以克服微生物培养限制的宏基因组技术,这为人们认识天然环境中蛋白质序列空间(protein universe)提供了可能。人们对宏基因组数据集进行初步分析就发现了数以千计的蛋白质新家族,并且家族内生物大分子序列数据量也在急剧增加[5]。然而,面对宏基因组产生的海量大数据,人们不可能对其每一条序列进行详尽的功能验证,对于多功能性的酶类也没有有效的实验技术为每一条序列进行全部底物与反应性质的验证,这就给新时代酶学研究提出了新的挑战。
碳水化合物酶是一类重要的活性蛋白,在宏基因组学快速发展之下,这类酶的研究和应用显得越来越重要。因此,本文中笔者综述了碳水化合物酶的研究背景、分类方法及其研究成果,以期为其在工业微生物领域的应用奠定基础。
1 碳水化合物酶类分类法研究
早在1989年,Henrissat等[6]基于疏水簇分析将21种β-聚糖酶类氨基酸序列进行比对,并根据氨基酸序列相似性划分成了6个纤维素酶家族。1991年,Henrissat又根据SWISS-PROT和EMBL/GenBank数据库中的氨基酸序列,基于蛋白质催化结构域的氨基酸序列相似性,对当时301种不同来源的糖苷水解酶类(glycoside hydrolases,GHs)序列进行分类[7],并不断进行更新[8-9]。这种分类系统的理论基础是氨基酸序列的相似性反映蛋白质保守的结构折叠类型。功能未知的氨基酸序列,可根据其序列相似性将其归类,形成特定的GH蛋白质家族。据此,不仅可以将不同糖苷水解酶进行分类,基于家族内序列相似性还可以分析其分子进化关系。这种分类方式随后扩展到糖基转移酶类(glycosyl transferases,GTs)[10]。随着碳水化合物酶类三维结构的获得,1997年该分类法中又加入蛋白质空间结构的信息,并基于催化结构域氨基酸序列与三维结构对碳水化合物酶家族进行重新分类[3]。以上这些可合成或分解碳水化合物的酶类,统称为CAZymes。1998年9月,CAZymes的这种分类正式在网络上开放,形成了专门的CAZy数据库(http:∥www.cazy.org/)。随后,人们发现自然界还存在部分没有催化活性即可辅助多糖降解酶进行降解的模块[11-12],最初报道的多为结合不溶纤维素、几丁质与淀粉等物质的模块。Warren及其同事研究发现,这些多糖结合模块也可形成特定家族——碳水化合物结合结构域(carbohydrate-binding modules,CBM)家族[13-15],这些家族及多糖裂解酶、碳水化合物酯酶也被CAZy列出并不断更新。
基于以上分类方法,Henrissat等[16]在1998年提出了一种全新的糖苷水解酶类命名方法,利用3个字母表示酶分子相关的底物,其后的数字表示所属的糖苷水解酶类家族,最后的大写字母表示该酶第一次报道时所排次序。后来,在不同物种中发现同功酶数目的增多,又在原先命名的基础上增加生物属名与种名首字母加入到命名当中。如Trichoderma reesei(瑞氏木霉)中的3种酶:CBHⅠ(纤维二糖水解酶Ⅰ)、CBHⅡ(纤维二糖水解酶Ⅱ)和EGⅠ(内切葡聚糖酶I)的催化结构域分别命名为TrCel7A,TrCel6A,TrCel6B。更多代表性的糖苷水解酶名称见表1。现在多数研究者们已经广泛采用这种命名系统,但仍有部分研究者未完全采用这种命名方法,主要原因是该命名未反映酶分子的底物专一性与降解模式,如该方法不能区分内切纤维素酶与外切纤维素酶等[17]。
最近研究发现,CBM33家族、GH61家族的部分组分其真实功能是裂解多糖单加氧酶类(lytic polysaccharide monooxygenases,LPMO),这是一类全新的氧化酶类,因此就需要对CBM与GH家族等相关家族重新进行分类[18]。另外,由于绿色植物细胞壁中木质素总与多糖类物质同时出现,并且降解木质素的酶类很可能与LPMO一同发生作用,因此CAZy数据库将降解木质素的酶类列入LPMOs家族,并创建一种全新的 CAZy大类,命名为辅助酶类家族[18](auxiliary activities,AAs),这样 CAZy数据库基本涵盖了木质纤维素降解所需要的相关酶类。截至2013年10月1日,CAZy数据库已经包含糖苷水解酶类(GHs)、糖苷转移酶类(GTs)、多糖裂解酶类(polysaccharide lyases,PLs)、糖水化合物酯酶类(carbohydrate esterases,CEs)、碳水化合物结合模块(CBMs)和辅助模块酶类(AAs)六大类家族,其家族数目分别达到了132、94、22、16、66 和10 个。
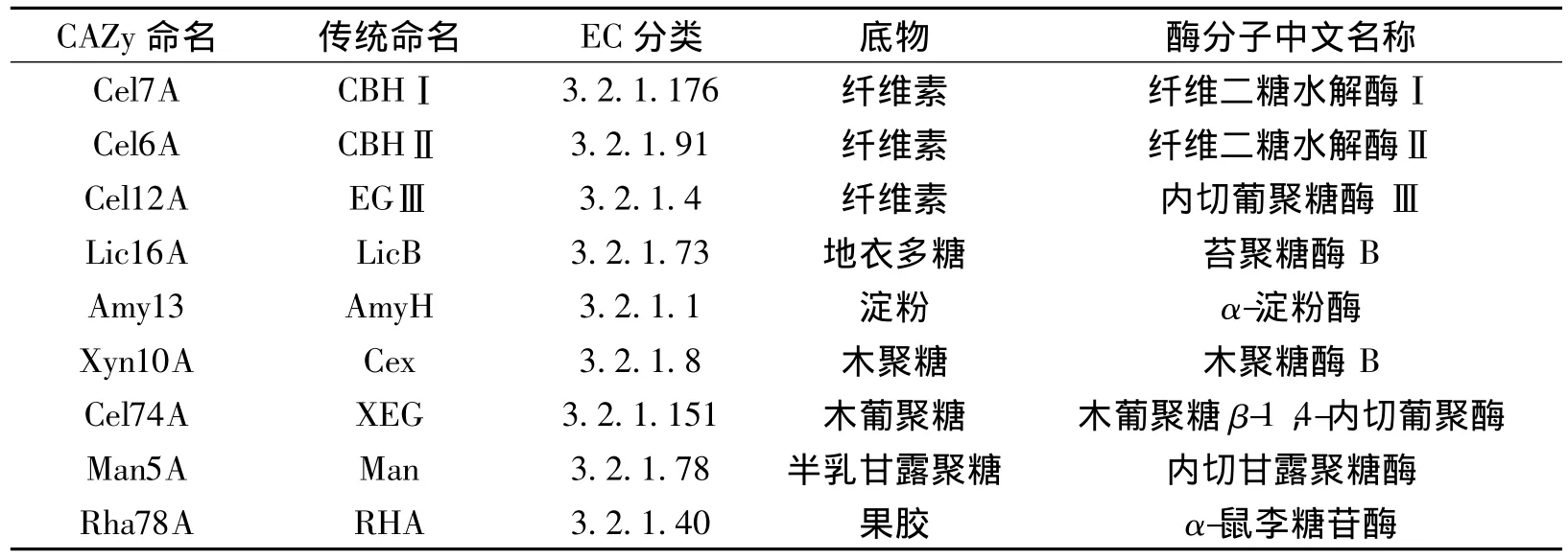
表1 CAZy糖苷水解酶命名方式Table 1 Designations for glucoside hydrolase enzymes
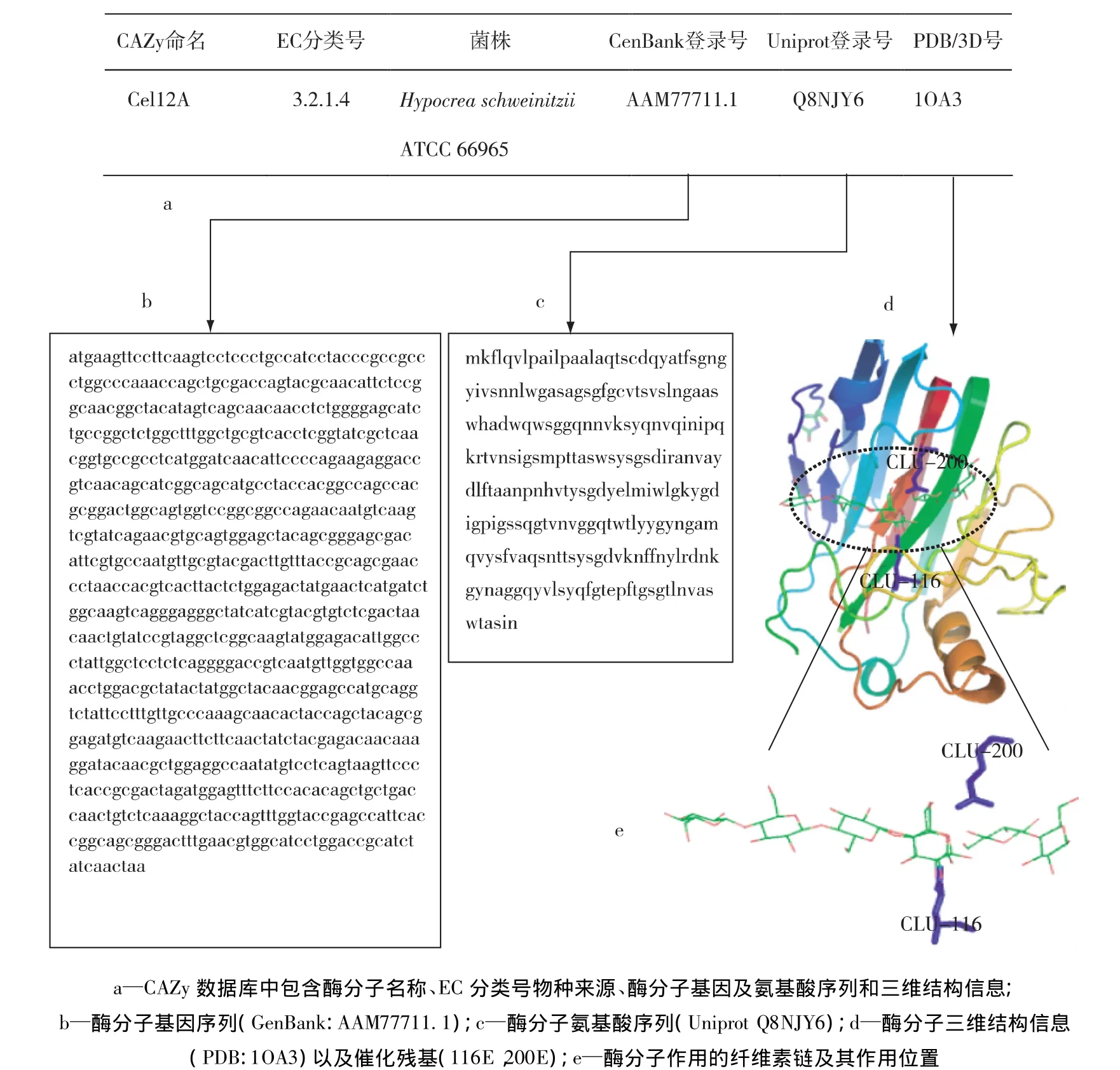
图1 CAZy数据库家族内列出酶分子相关信息(以AAM77711.1为例)Fig.1 Information of enzymes contained in CAZy database(AAM77711.1)
2 CAZy数据库——基因组学与酶学研究的重要桥梁
CAZy数据库中列出了酶分子序列的家族信息、物种来源、基因序列、蛋白质序列信息、三维结构、EC分类以及与相关数据库的链接。对于每一家族中已经得到生化表征的酶分子,还提供催化机制关系密切的信息,包括活性中心及催化机制特征,催化残基(对整个家族是保守的)及其分类范围信息,这些信息对快速分析同一家族酶分子共同特征是非常重要的。图1以AAM77711.1为例显示了CAZy数据库家族内列出的酶分子相关信息。
CAZy数据库建立的目的是将酶分子的序列、结构与催化机制特点结合起来,对其结构域进行定义。碳水化合物活性酶类常常是多结构域的,在CAZy数据库中,同一条基因不同结构域划入不同的结构域家族,如T.reesei分泌的CBHⅠ碳水化合物结合模块归入CBM1家族,而催化结构域归入GH7家族[19]。这样对包含多个结构域的酶分子定义更加准确,特别是研究复杂的木质纤维素高效降解生境系统,通过研究酶分子基因结构域的组合就可以了解相应微生物的降解模式与降解策略。如东秀珠课题组利用宏基因组技术研究牦牛瘤胃降解植物细胞壁酶的多样性时发现[20],降解纤维素的基因在宏基因组序列中含量丰富,从其构建的开放阅读框(ORFs)中分析蛋白质结构域,发现具有水解酶酶活力的蛋白质来自GH5、9、10等糖苷水解酶家族,并且这样的结构域与编码SusC/SusD类型的外膜蛋白结构域相连,只有少量催化结构域带有碳水化合物结合模块,没有检测到催化结构域与纤维小体的对接/粘连模块相连。这些发现表明,在牦牛瘤胃木质纤维素降解过程中起着重要作用的纤维素酶类应与SucC/SucD有关的催化机制,明显不同于热纤梭菌采用的纤维素小体模式,也不同于丝状真菌大量分泌胞外游离酶系的模式[20]。
利用生物信息学手段快速筛选由宏基因组产生的大量基因序列,确定相关基因功能结构域的组合方式,可以预测相关微生物采取的降解模式是属于游离酶系(只有催化结构域或包含催化结构和CBM模块)、纤维小体超分子复合物(含有对接/粘连模块、锚定模块等)还是其他模式[21-23](图2),这就大大降低了实验的工作强度,明确了研究目标并具有一定针对性。将相关功能结构域归入某一蛋白家族后,由于家族内蛋白质的三维结构非常保守[3,24],催化机制也非常保守,确定其相关蛋白质家族后,GH家族的酶分子序列就可以确定其催化机制是保留型还是反转型。如果该蛋白质家族中有一酶组分的三维结构获得解析,人们还可以利用同源模建技术获得相应酶分子的结构特性,尤其是催化活性中心及其催化活性位点附近的空间信息,这就大大提高了酶分子结构与功能研究的工作效率[1]。
3 CAZy数据库的新研究趋势
由于测序技术的飞速发展,宏基因组研究产生了海量的生物多样性与序列多样性数据,现在蛋白质序列的发现速度已经远远超出人们对功能确切描述与分析的速度[22]。如CAZy数据库的蛋白序列已经达到34万余条(截至2013年10月1日),获得生化表征序列却仅有1万余条,不足3%;而获得三维结构的序列仅有 1 400多个,不足 0.5%。CAZy数据库现在面临的难题可能不再是蛋白质序列太少,而是如何对宏基因组产生的大数据(big data)进行深入地挖掘分析。
宏基因组技术产生海量数据,人们不再可能穷举所有序列、所有底物,逐条逐项地分析其生物学功能,必须运用生物信息学方法建立相关算法,完成其自动功能注释[25-26]。早在人类基因组草图完成时,有人就利用同源性方法来预测蛋白质的功能,提出了结构基因组学(structural genomics)的概念,以序列一致性30%为标准构建蛋白质家族,利用同源模建方法来分析其结构与功能[27]。然而,由于序列同源性并不意味着蛋白质具有相同的功能,不同基因由于处于不同选择压力之下,因而可能具有不同的进化速率,这使得预测结果的准确性难以确定[28]。特别是酶分子功能执行区域仅是催化结构域中非常小的一部分,仅仅基于全序列比对结果来预测局部发生变化区域的功能,这是自动功能注释常常出错的根源[29]。对应酶分子功能分类层次,如EC号包括4级层次:酶的大类、化学键类型、反应类型及底物专一性。蛋白质家族分类也应根据相似性程度进行不同层次的聚类分析,以对应酶分子功能分类的不同层次,来提高预测的效率与准确度[30]。现在CATH等蛋白质结构分类数据库已经根据不同的序列一致性细化出不同的层次,序列一致性<35%为S层,<60%为O层,<95%为L层,100%为I层[31]。Pfam等蛋白质数据库也加强了与架构保守性(即功能位点保守性)数据库如Prosite、SCOP和CAZy等的联系,以提高其功能预测的准确性[32]。
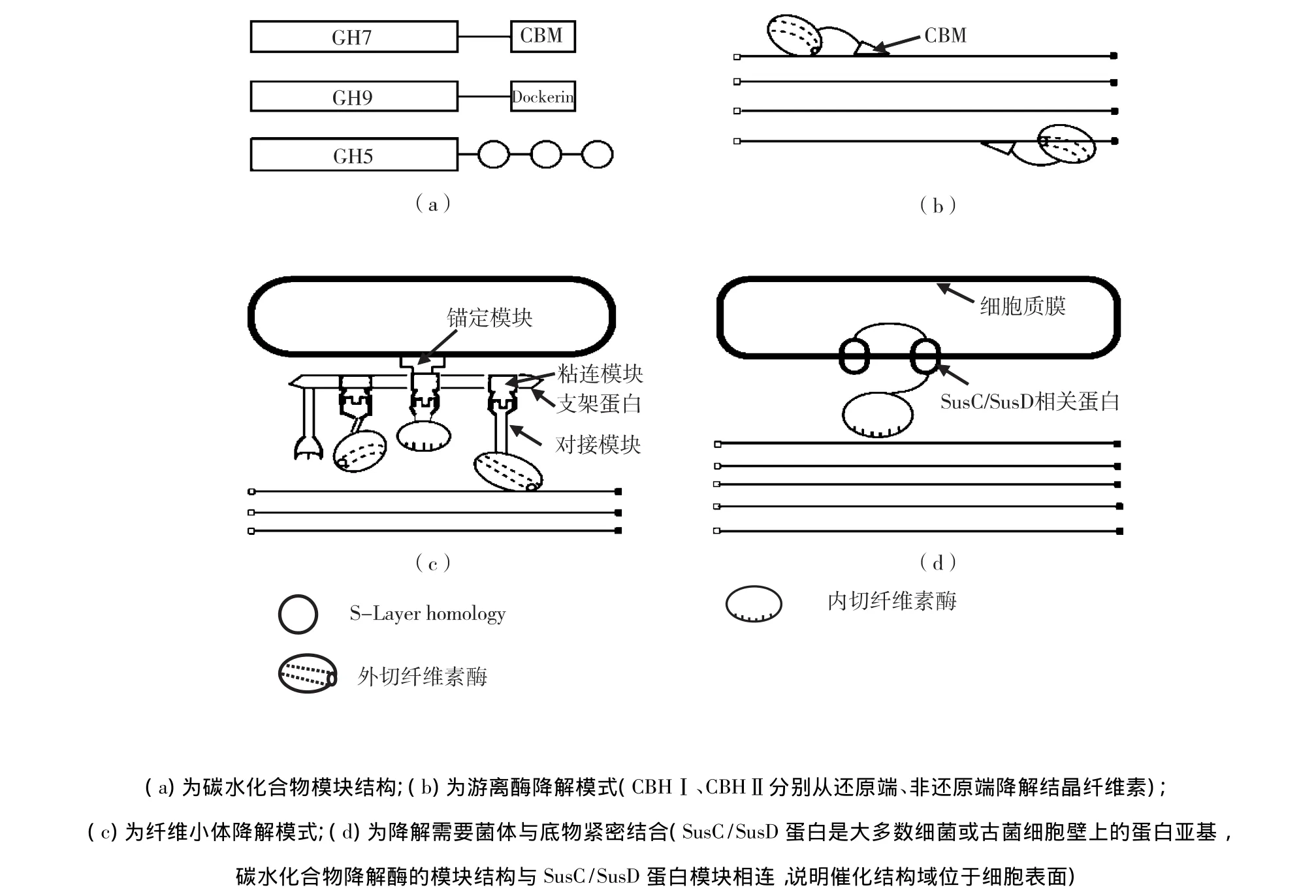
图2 碳水化合物降解酶的模块结构及其可能的降解模式Fig.2 Domain architectures and its possible mode of degradation
CAZy数据库对酶分子催化结构域按30%序列相似性进行家族分类,不能够准确预测同一家族内不同成员的底物专一性。随着宏基因组数据的快速增加,CAZy数据库也正在着手对所包含家族进行细化分类。其中糖苷水解酶类(GHs)涵盖CAZy数据库中最多的家族,是CAZy数据库中生化特征被描述最为详细的酶类。目前,CAZy数据库已经对糖苷水解酶GH5、GH13和GH30家族进行了亚家族的分类[33-34]。以 GH5家族为例,GH5家族是CAZy库中最大的一个糖苷水解酶家族,因为它是第1个纤维素酶家族,该家族曾被命名为“纤维素酶家族A”[6]。GH5家族序列分布很广,在古菌、细菌和真菌界(真菌、植物)都存在,利用宏基因组学方法从不同生境中也鉴定出了丰富的GH5家族序列[35-37]。GH5模块的折叠类型是 TIM结构,实验确定了近20种明确EC分类的酶活性,这充分展现了该家族的多功能性。因此仅将蛋白质序列归入如此庞大的“多专一性”家族显然不能够发掘出依靠序列与结构相似性进行分类的全部潜力,基于序列一致性>75%的标准,提出了GH5家族新的亚家族分类系统,其中51个亚家族能覆盖其中80%以上的序列[33]。
经过进一步的功能分析之后,发现GH5家族中有的亚家族(表2)是单底物专一性的亚家族,如GH5-5、GH5-8亚家族等,对那些多功能的亚家族再进行细化分类就可能形成单功能亚亚家族,当新发现的序列归入此类亚家族或亚亚家族时,就可以判断该序列可能具有此类功能,这有利于提高功能注释的准确度与效率。而多功能性亚家族或亚亚家族序列相似性很高,这说明其中只要几个氨基酸的突变就可能导致功能的分歧[33],对其中GH5-4亚家族的改造也证明了这一点,他们利用全面的GH5亚家族系统发育分析发现了GH5-4亚家族中决定葡聚糖和甘露聚糖双底物特异性活性位点的基序[38]。

表2 GH5家族各亚家族底物专一性(以GH5-1到GH5-10亚家族为例)Table 2 Subfamilies with identified active enzymes in GH5(subfamily GH5-1 to GH5-10 as example)
表2列出的GH5家族8个亚家族中,GH5-5、GH5-8亚家族具有单底物专一性;其余4个亚家族均具有两个或两个以上多底物专一性(GH5亚家族A1-A10是较先发现的亚家族,在进行重新分类时,A3归入GH5-4亚家族,A5和A6统一归入到GH5-5亚家族,为了与先前的分类一致),这些重新划分的亚家族保持了与原先一样的序号[39]。
4 展望
随着测序技术的进一步发展,宏基因组技术产生的海量蛋白质序列既是挑战,又是机遇。CAZy数据库将碳水化合物酶类序列归入不同的“多专一性”家族,通过对蛋白质家族分类的进一步细化,对亚家族甚至亚亚家族的分类,找到更小的聚类族,分析与酶分子功能密切相关的活性中心部位,确定酶分子决定功能专一性残基/组合及其协变性,就可以提高功能预测的准确度,这对于了解碳水化合物活性酶类的作用机制具有重要意义。对碳水化合物活性酶类亚家族、亚亚家族的分类,使得同一亚家族或亚亚家族内氨基酸序列相似性很高,几个氨基酸的改变就可能改变酶的功能,这就大大降低了蛋白质工程改造对序列空间的搜索强度,提高了理性设计成功的概率[40],对生物质转化和生物炼制提供了有力的技术支持。同时,这种亚家族的分类方法对其他类型蛋白质的功能预测也具有重要指导意义。
[1] Cantarel B L,Coutinho P M,Rancurel C,et al.The carbohydrateactive enzymes database(CAZy):anexpertresourcefor glycogenomics[J].Nucleic Acids Res,2009,37:D233-D238.
[2] HultK,Berglund P.Enzymepromiscuity:mechanism and applications[J].Trends Biotechnol,2007,25(5):231-238.
[3] Henrissat B, Davies G.Structural and sequence-based classification of glycoside hydrolases[J].Curr Opin Struct Biol,1997,7(5):637-644.
[4] Vlasenko E,Schülein M,Cherry J,et al.Substrate specificity of family 5,6,7,9,12,and 45 endoglucanases[J].Bioresour Technol,2010,101(7):2405-2411.
[5] Godzik A.Metagenomics and the protein universe[J].Curr Opin Struct Biol,2011,21(3):398-403.
[6] Henrissat B,Claeyssens M,Tomme P,et al.Cellulase families revealed by hydrophobic cluster analysis[J].Gene,1989,81(1):83-95.
[7] Henrissat B.A classification of glycosyl hydrolases based on amino acid sequence similarities[J].Biochem J,1991,280:309-316.
[8] Henrissat B,Bairoch A.New families in the classification of glycosyl hydrolases based on amino acid sequence similarities[J].Biochem J,1993,293:781-788.
[9] Henrissat B, Bairoch A.Updating the sequence-based classification of glycosyl hydrolases[J].Biochem J,1996,316:695-696.
[10] Campbell J A,Davies G J,Bulone V,et al.A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities[J].Biochem J,1997,326:929-942.
[11] Svensson B,Jespersen H,Sierks M R,et al.Sequence homology between putative raw-starch binding domains from different starch-degrading enzymes[J].Biochem J,1989,264:309-311.
[12] Gilkes N R,Henrissat B,Kilburn D G,et al.Domains in microbial beta-1,4-glycanases:sequence conservation,function,and enzyme families[J].Microbiol Rev,1991,55(2):303-315.
[13] Coutinho J B,Gilkes N R,Kilburn D G,et al.The nature of the cellulose-binding domain effects the activities of a bacterial endoglucanase on different forms of cellulose[J].FEMS Microbiol Lett,1993,113(2):211-217.
[14] Tomme P,Warren R A J,Miller R C,et al.Cellulose-binding domains:classification and properties[C].ACS Symposium Series,1995,618:142-163.
[15] Warren R A J.Microbial hydrolysis of polysaccharides[J].Ann Rev Microbiol,1996,50(1):183-212.
[16] Henrissat B,Teeri T T,Warren R A J.A scheme for designating enzymes that hydrolyse the polysaccharides in the cell walls of plants[J].FEBS Lett,1998,425(2):352-354.
[17] 曲音波,陈冠军,高培基,等.木质纤维素降解酶与生物炼制[M].北京:化学工业出版社,2011.
[18] Levasseur A,Drula E,Lombard V,et al.Expansion of the enzymatic repertoire of the CAZy database to integrate auxiliary redox enzymes[J].Biotechnol Biofuels,2013,6(1):1-14.
[19] Sukharnikov L O,Cantwell B J,Podar M,et al.Cellulases:ambiguous nonhomologous enzymes in a genomic perspective[J].Trends Biotechnol,2011,29(10):473-479.
[20] Dai X,Zhu Y,Luo Y,et al.Metagenomic insights into the fibrolytic microbiome in yak rumen[J].PLoS One,2012,7(7):e40430.
[21] Wilson D B.Microbial diversity of cellulose hydrolysis[J].Curr Opin Microbiol,2011,14(3):259-263.
[22] Wilson D B.Processive and nonprocessive cellulases for biofuel production:lessons from bacterial genomes and structural analysis[J].Appl Microbiol Biotechnol,2012,93(2):497-502.
[23] Medie F M,Davies G J,Drancourt M,et al.Genome analyses highlight the different biological roles of cellulases[J].Nat Rev Microbiol,2012,10(3):227-234.
[24] Davies G,Henrissat B.Structures and mechanisms of glycosyl hydrolases[J].Structure,1995,3(9):853-859.
[25] Ferrer M,Beloqui A,Timmis K N,et al.Metagenomics for mining new genetic resources of microbial communities[J].J Mol Microbiol Biotechnol,2008,16(1/2):109-123.
[26] Friedberg I.Automated protein function prediction:the genomic challenge[J].Brief Bioinform,2006,7(3):225-242.
[27] Baker D,Sali A.Protein structure prediction and structural genomics[J].Science,2001,294(5540):93-96.
[28] Lee D,Redfern O,Orengo C.Predicting protein function from sequence and structure[J].Nat Rev Mol Cell Biol,2007,8(12):995-1005.
[29] Sjölander K.Getting started in structural phylogenomics[J].PLoS Comput Biol,2010,6(1):e1000621.
[30] Prakash T,Taylor T D.Functional assignment of metagenomic data:challenges and applications[J].Brief Bioinform,2012,13(6):711-727.
[31] Sillitoe I,Cuff A L,Dessailly B H,et al.New functional families(FunFams)in CATH to improve the mapping of conserved functional sites to 3D structures[J].Nucleic Acids Res,2013,41(D1):D490-D498.
[32] Bateman A,Coin L,Durbin R,et al.The Pfam protein families database[J].Nucleic Acids Res,2004,32(S1):D138-D141.
[33] Aspeborg H,Coutinho P M,Wang Y,et al.Evolution,substrate specificity and subfamily classification of glycoside hydrolase family 5(GH5)[J].BMC Evol Biol,2012,12(1):186.doi:10.1186/1471-2148-12-186.
[34] Stam M R,Danchin E G,Rancurel C,et al.Dividing the large glycoside hydrolase family 13 into subfamilies:towards improved functional annotationsofalpha-amylase-related proteins[J].Protein Eng Des Sel,2006,19(12):555-562.
[35] Duan C J,Xian L,Zhao G C,et al.Isolation and partial characterization of novel genes encoding acidic cellulases from metagenomes of buffalo rumens[J].J Appl Microbiol,2009,107(1):245-256.
[36] Elifantz H,Waidner L A,Michelou V K,et al.Diversity and abundance of glycosyl hydrolase family 5 in the North Atlantic Ocean[J].FEMS Microbiol Lett,2008,63(3):316-327.
[37] Hess M,Sczyrba A,Egan R,et al.Metagenomic discovery of biomass-degrading genes and genomes from cow rumen[J].Science,2011,331:463-467.
[38] Chen Z,Friedland G D,Pereira J H,et al.Tracing determinants of dual substrate specificity in glycoside hydrolase family 5[J].J Biol Chem,2012,287(30):25335-25343.
[39] Lo L L,Larsen S.The 1.62 Å structureofThermoascus aurantiacus endoglucanase:completing the structural picture of subfamilies in glycoside hydrolase family 5[J].FEBS Lett,2002,523(1/2/3):103-108.
[40] Lichtarge O,Wilkins A.Evolution:a guide to perturb protein function and networks[J].Curr Opin Struct Biol,2010,20(3):351-359.
