Properties of Klebsiella Phage P13 and Associated Exopolysaccharide Depolymerase
2014-05-02LIUYangLIGuiyangMOZhaolanCHAIZihanSHANGAnqiandMOUHaijin
LIU Yang, LI Guiyang, MO Zhaolan, CHAI Zihan, SHANG Anqi, and MOU Haijin,
1) College of Food Science and Engineering, Ocean University of China, Qingdao 266003, P. R. China
2) Yellow Sea Fishery Research Institute, Chinese Academy of Fishery Sciences, Qingdao 266071, P. R. China
Properties of Klebsiella Phage P13 and Associated Exopolysaccharide Depolymerase
LIU Yang1), LI Guiyang2), MO Zhaolan2), CHAI Zihan1), SHANG Anqi1), and MOU Haijin1),*
1) College of Food Science and Engineering, Ocean University of China, Qingdao 266003, P. R. China
2) Yellow Sea Fishery Research Institute, Chinese Academy of Fishery Sciences, Qingdao 266071, P. R. China
The bacteriophage P13 that infectsKlebsiellaserotype K13 contains a heat-stable depolymerase capable of effective degradation of exopolysaccharide (EPS) produced by this microorganism. In this study, the titer of phage P13, initially 2.0 × 107pfu mL−1, was found increasing 20 min after infection and reached 5.0 × 109pfu mL−1in 60 min. Accordingly, the enzyme activity of depolymerase approached the maximum 60 min after infection. Treatment at 70℃ for 30 min inactivated all the phage, but retained over 90% of the depolymerase activity. Addition of acetone into the crude phage lysate led to precipitation of the protein, with a marked increase in bacterial EPS degradation activity and a rapid drop in the titer of phage. After partial purification by acetone precipitation and ultrafiltration centrifugation, the enzyme was separated from the phage particles, showing two components with enzyme activity on Q-Sepharose Fast Flow. The soluble enzyme had an optimum degradation activity at 60℃ and pH 6.5. Transmission electron microscopy demonstrated that the phage P13 particles were spherical with a diameter of 50 nm and a short stumpy tail. It was a double-strand DNA virus consisting of a nucleic acid molecule of 45976 bp. This work provides an efficient purification operation including thermal treatment and ultrafiltration centrifugation, to dissociate depolymerase from phage particles. The characterization of phage P13 and associated EPS depolymerase is beneficial for further application of this enzyme.
Klebsiella; bacteriophage; exopolysaccharide; depolymerase; enzymatic hydrolysis
1 Introduction
To protect cells against environmental stresses, bacteria tend to produce various extracellular substances and form bacterial biofilm in a broad range of environments, especially in food processing fields. Major components of such bacterial biofilm include water, exopolysaccharides (EPS), proteins, lipids, mineral ions and cells (Flintet al., 1997; Coenye and Nelis, 2010). To date, more than 80 kinds of EPS with different chemical compositions have been reported inKlebsiella, which surround the cell surface and connect cells to form the biofilm with complicated structure (Griffiths and Davies, 1991; Sutherland, 2001). A variety of techniques with chemical disinfectants and antiseptics have successfully been used to degrade the undesired biofilm. However, their side effects should not be ignored. During the treatments, resistance may be induced in some cells by mutation or gene transfer for survival and growth (Davies, 2003). The bacteriophageborne depolymerase, an enzyme specifically degrading bacterial EPS, is potentially effective for controlling microorganisms involved in biofilm formation (Stewartet al., 1995). The bacterial EPS-degrading ability of bacteriophage-borne EPS depolymerase has been recorded for more than 50 years (Simoeset al., 2010). The enzyme is released by bacteriophage during its infection and binding to the capsular material of bacterial cells. Then, the capsular polymer is degraded and the phage is allowed to access bacterial cell, further binding to an outer-membrane receptor (Hugheset al., 1998). There has been a study using phage-borne glycanase to facilitate the removal of biofilm from clinical settings (Hanlonet al., 1998). This enzyme has also been used for other purposes such as biological control against animal and plant pathogens (Kim and Geider, 2000; Scorpioet al., 2008), preparation of novel oligosaccharides with potential biological activities (Duttonet al., 1981; Di Fabioet al., 1985), and structure analysis of bacterial EPS (Altmannet al., 1990; Cescuttiet al., 1993).
The properties of bacteriophage-borne depolymerase have rarely been reported. The enzyme randomly attacks the glycosidic bond of capsular polysaccharide to release repeating units of the polymer. It normally occurs in two forms,i.e., soluble and phage-bound depolymerase, with similar enzymatic and other properties. The presence of soluble enzyme may indicate the overproduction of gene product from the phage genome (Yurewiczet al., 1971). The illumination of reaction mechanisms and enzymological properties of bacteriophage-borne depolymerase en-zymes is necessary for further application of the enzyme. In this paper, a heat-stable phage-associated depolymerase with high-performance of EPS hydrolyzation was isolated fromKlebsiellaK13. In addition, the properties of the phage and associated depolymerase were characterized.
2 Materials and Methods
2.1 Strains
The bacteriophage P13 capable of infectingKlebsiellaserotype K13 (NCTC 9133) was isolated from sewage samples according to the method reported by Mouet al.(2008).
2.2 Preparation of EPS
KlebsiellaK13 was grown in nutrient broth at 32℃ for 24 h, and then transferred to 1500 mL of fresh fermentation medium with the following composition (g L−1): CaCl2·2H2O, 0.001; Na2HPO4, 10; KH2PO4, 3; NaCl, 1; MgSO4·7H2O, 0.2; FeSO4·7H2O, 0.0001; K2SO4, 1; glucose, 30; beef extract, 0.5; peptone, 0.5; and oleic oil, 1.5. After 48 h incubation at 24℃, the fermentation liquor was centrifuged and the supernatant was concentrated by rotary evaporation at 60℃ to approximately one-third of the initial volume. Then three-fold volume of 95% icy ethanol was added for EPS sedimentation. The precipitate was dissolved in distilled water, sealed in dialysis bags (cut-off value, 12000–14000 Da), and dialyzed against distilled water at 4℃ for 48 h. Sodium azide was added to the EPS for long-term preservation.
2.3 Preparation of Crude Depolymerase
KlebsiellaK13 was cultured in nutrient broth at 32℃in a shaking incubator at 150 r min−1until the optical density at 600 nm (OD600) reached 0.4 (cell density 3× 1010–4 × 1010cfu mL−1). The culture was infected with phage suspension in a volume ratio of 10:1. The incubation was continued under the similar conditions until OD600was below 0.1. The lysate was processed immediately for purification by centrifugation at 8000 r min−1and 4℃ for 10 min. The supernatant was filtered (Millipore, 0.22 μm pore size) into a sterile screw cap bottle to remove surviving bacterial cells or cell debris. The filtered phage suspension was dialyzed (cut-off value, 12000–14000 Da) against distilled water for 48 h at 4℃ to remove the residual broth components, and the retentate was taken as the crude enzyme extract solution.
Icy acetone (4℃) was added slowly under stirring to the crude enzyme extract solution in a volume ratio of 3:2 and the mixture was incubated at 4℃ for 24 h to allow protein precipitation. The precipitate was collected by centrifugation and re-dissolved with ultrapure water. The re-dissolved solution was dialyzed against distilled water at 4℃ for 48 h to remove the low molecular mass materials including acetone. The retentate was freeze-dried and enzyme powder was obtained.
2.4 Ultrafiltration of Enzyme
The enzyme powder was re-dissolved in ultrapure water at a final concentration of 1 mg mL−1. The aqueous fluid was separated using Sartorius vivaspin (molecular weight cut-off of 100 kDa, 6 mL) by centrifugation at 3000 r min−1until the volume ratio of retentate to filtrate was approximately 3:7. Enzyme activity and phage titer were determined immediately.
2.5 Purification of Enzyme by Column Chromatography
The prepared enzyme solution was loaded onto a Q-Sepharose Fast Flow column (40 cm × 2.5 cm, Amersham-Pharmacia Biotech, Sweden) equilibrated with 0.2 mol L−1, pH 6.5 phosphate buffer solution. The flow rate was adjusted to 2 mL min−1. The protein was eluted from the column with the same buffer containing a linear NaCl gradient (0–2.0 mol L−1). The elute fractions were continuously monitored using a UV detector at 280 nm and collected for enzyme activity assays against bacterial EPS.
SDS-PAGE of depolymerase was performed on 6% stacking gel and 12% separation gel at 120 V according to the method of Zhu (2010). The protein bands were visualized by staining the gel with Coomassie blue.
2.6 Determination of Enzyme Activity and Phage Titer
The EPS-degrading activity of bacteriophage crude depolymerase was assayed by determination of the increase in reducing sugar levels as described by von Borelet al. (1952). Two mL of 0.5% EPS was mixed with 0.4 mL of filtered crude phage lysate and then incubated at 60℃for 30 min. Then, 1.5 mL of reaction solution was mixed with 1.5 mL of 3, 5-dinitrosalicylic acid (DNS), heated at 100℃ for 5 min, cooled to room temperature and diluted to 25 mL with deionized water. The optical density was read at 520 nm and values for reducing sugars were expressed as D-glucose equivalents. One unit of phage enzyme activity was defined as the amount of enzyme that released 1 μg of D-glucose per min under the above conditions.
Sloppy agar (0.6% nutrient agar, Sigma) was used for titer counting of phage. Equal volumes of bacterial cells and phage suspension (0.1 mL) were mixed with 3 mL of melted sloppy agar, and poured on a nutrient agar plate. After incubation at 32℃ for 48 h, the number of phage particles on the plates was enumerated as the resulting plaque-forming units (pfu).
Total protein content was determined by Coomassie brilliant blue assay (Xuet al., 2010).
2.7 Effects of Environmental Factors on Enzyme Activity
The optimal pH was determined by measuring the enzyme activity under different pH conditions (5.5–8.0) at 60℃. The optimal temperature was determined by measuring the enzyme activity at pH 6.5 and temperatures ranging from 10℃ to 70℃. To determine the heat-stability, the enzyme was pretreated at different temperaturesfor 30 min or 2 h and then assayed for enzyme activity at 60℃ using EPS ofKlebsiellaK13 as the substrate. To analyze the effects of metal cations on enzyme activity, phage enzyme was incubated with EPS in the presence of different metal cations (CaCl2, KCl, NaCl and MgSO4·7H2O) at a final concentration of 0.1 mol L−1. In addition, the effect of EDTA on the enzyme activity was studied using the same method.
2.8 Extraction of Phage Nucleic Acid
The phage suspension was sealed in dialysis bags (cut-off value, 12000–14000 Da) and 20-fold concentrated by PEG 20000. The residual host cells and contaminating microorganisms were removed by filtration (0.22 μm pore size, Millipore, USA). DNase І (D5025, Sigma, USA) and RNase A (EN05311, Fermentas, USA) were added to the filtrate at a final concentration of 0.25 U μL−1and 0.2 μg μL−1, respectively, for digestion of DNA and RNA released by host cells during the infection of phage. After the removal of nuclease, phage nucleic acid was extracted from the phage particles by UNIQ-10 genome extraction kit (Sangon, Shanghai, China). The purity of phage nucleic acid was evaluated by restriction enzymeNdeІ digestion, which made a distinction between phage P13 andKlebsiellaK13 nucleic acid.
2.9 Transmission Electron Microscopy (TEM) of Phage Particles
For TEM analysis, the phage suspension was concentrated by PEG 20 000, and the phage particles were collected by ultracentrifugation at 83000×g for 3 h (CP70ME, Hitachi, Japan). The phage suspension was fixed by 3% glutaraldehyde for 10 min. A drop of the phage fixation liquid was placed on a grid coated with a defatted carbon-holey-film and aired at natural state for 5 min. The excess liquid was withdrawn with a filter paper (Bayer and Anderson, 1963). The sample was stained with a drop of 1% phosphotungstic acid for 3 min with excess liquid removed with filter paper. The grid was introduced into a JEM-1200EX transmission electron microscope (JEOL, Japan) with an acceleration voltage of 80 kV. The phage particles with typical morphological characteristics were recorded under 10 k× to 180 k×.
3 Results and Discussion
3.1 Preparation of Bacterial EPS
Bacterial EPS commonly contains repeating single units joined by glycosidic linkages, mostly with the sequence of two to eight monosaccharides and unusual sugar residues. These can be homo- or heteropolymers substituted by both organic and inorganic molecules (Roberts, 1996). For example, theKlebsiellagenus has been classified into more than 80 serotypes on the basis of serological reactions with K-antigen (Griffithset al., 1991). In order to improve the EPS yield byKlebsiellaK13, single factor and the orthogonal test were used here to optimize the fermentation conditions. The addition of 0.2% Tween 80 or 0.15% oleinic acid was beneficial for increasing the EPS yield. After incubation in the optimum fermentation medium at 24℃ for 48 h, the yield of bacterial EPS reached 6.7 g L−1.
3.2 Fluctuating Curve of Phage and Enzyme During Infection
At the beginning of phage infection, the depolymerase released from phage P13 showed a rapid increase in its EPS-degrading activity, which promoted the invasion of the phage particles. However, the titer of phage remained stable at approximately 2.0×107pfu mL−1in the first 20 min during infection, indicating that the latency time of phage P13 was approximately 20 min. Results of OD600measurement and plate counting showed that the K13 cell density remained at the level close to the initial value (Fig.1). Then, there was a significant correlation between the rapidly increasing titer of phage and decreasing number of K13 cells from 20 to 60 min. After 60 min, the quantity of phages and cells stabilized, whereas the phage gave a final titration of 4.3×109pfu mL−1at 100 min. The host cells reduced from the initial number of 3.2×1010cfu mL−1to 9.0×104cfu mL−1(3.6×105-fold reduction). Reducing sugar determination showed that the production of bacterial EPS depolymerase increased from 0.046 to 0.397 and approached the maximum at 60 min.
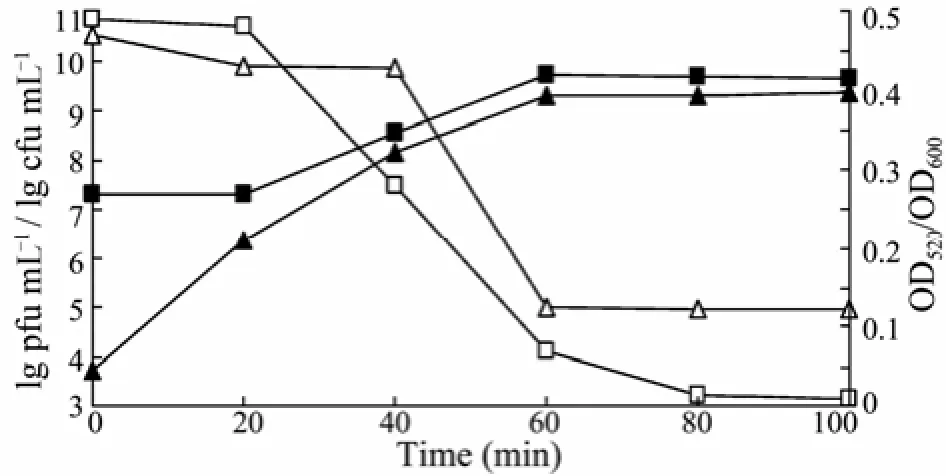
Fig.1 Changes in phage P13 titer and EPS depolymerase activity after phage infection. Cell density (□) was monitored at OD600. Remaining cells numbers (lg cfu mL−1) were counted on nutrient agar plate (△). Titer of phage (lg pfu mL−1) was determined according to the number of plaques formed on sloppy agar (■). Enzyme activity (▲) is represented as OD520using the DNS method.
3.3 Purification of Phage P13 Enzyme
The enzyme activity and phage titer were assayed after preliminary purification by acetone precipitation and ultrafiltration centrifugation (Table 1). The addition of acetone in a volume ratio of 3:2 (acetone: crude enzyme solution) largely inactivated the phage particles and EPS depolymerase, and enzyme activity was substantially low after acetone precipitation. When the supernatant collected from acetone precipitation was spot-inoculated onto nutrient plates spread with host cells, no phage plaque or halo was formed. Enzyme activity assays using the DNS method with bacterial EPS as the substrate confirmed the complete inactivation of enzyme in the supernatant. By comparison, approximately 13.8% of enzymeactivity was recovered in the precipitate. Only a small amount of active phage particles remained, and the total phage titer was decreased from 1.7×1011to 3×103pfu.
The material precipitated by acetone was collected with large molecular particles including phage particles removed via 100 kDa-ultrafiltration centrifugation. Phage particles with molecular weight greater than 100 kDa were not found in the filtrate. After the separation, approximately 34% of enzyme activity remained in the retentate and 66% of enzyme activity penetrated through the ultrafilter. These indicate that the phage depolymerase enzyme is a protein with a molecular weight less than 100 kDa. Purification of the enzyme from phage particles and other impurities by acetone precipitation and ultrafiltration centrifugation recovered 8.9% of the enzyme with the activity increased by 12.4-fold. It is possible that the phage enzyme was freely diffusible or loosely associated with the phage particles (Adams and Park, 1956).
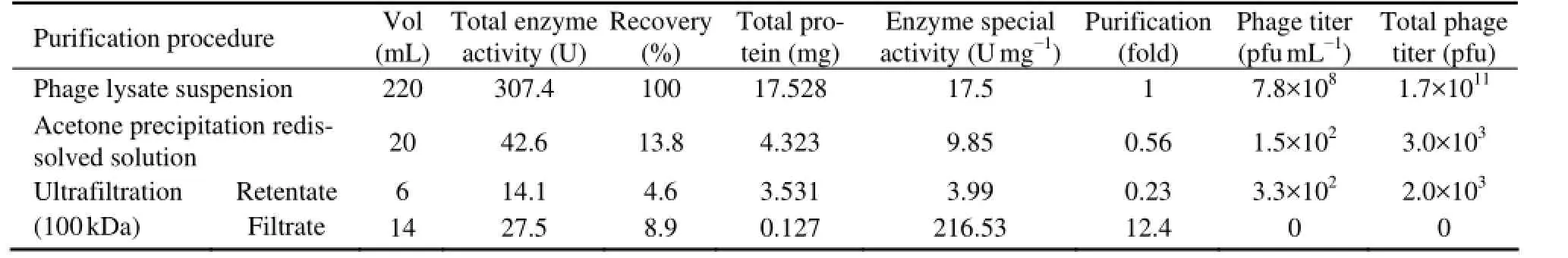
Table 1 Enzyme activity and phage titer of the bacteriophage-borne depolymerase during purification
The proteins precipitated by acetone showed several peaks on the eluting curve of Q-Sepharose Fast Flow (Fig.2). Two of them showed enzyme activities (E1 and E2). After ultrafiltration centrifugation, the filtrate showed only one peak with enzyme activity (E2) on the eluting curve (data not shown). This indicates that the phage P13 produced two different types of depolymerase, or the same depolymerase occurred in soluble and phagebound forms. The latter explanation could be supported by the evidence that the phage particles were larger than 100 kDa and were removed by ultrafiltration. The soluble enzyme is likely resulting from the overproduction by the phage genome (Yurewiczet al., 1971) and released by phage particles during lysis of host cells. SDS-PAGE assay showed that the molecular weight of phage P13 depolymerase is 62–65 kDa.
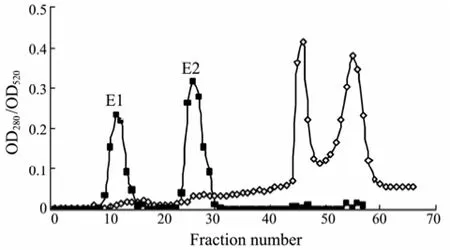
Fig.2 The elution curve of enzyme by Q-Sepharose Fast Flow. The sample was eluted with 0.2 mol L−1phosphate buffer solution having a linear NaCl gradient (0–2.0 mol L−1) at an eluant velocity of 2 mL min−1. The protein content (◊) was monitored by UV detector at 280 nm. The enzyme activity (■) was assayed based on the released reducing sugar level detected by the DNS method at 520 nm.
3.4 Properties of Phage P13 Enzyme
The addition of phage P13 rapidly decreased the viscosity of EPS solution within minutes, showing that phage EPS depolymerase is a high-efficiency endo-glycanohydrolase. Reduction in EPS viscosity might also aid bacteriophage to penetrate bacterial biofilm (Hanlonet al., 2001). Within the first few hours of hydrolysis, the content of reducing sugar produced by EPS hydrolysis increased rapidly with the reaction time. The OD520reached the maximum value in 4 h of hydrolysis.
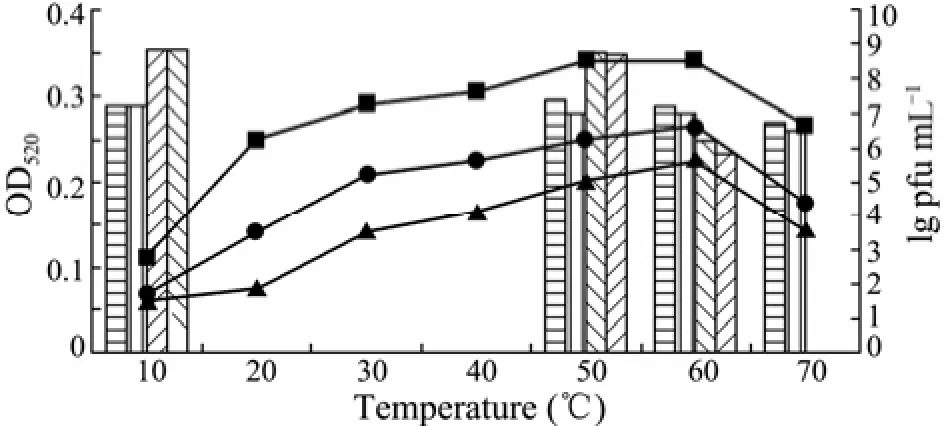
Fig.3 Effects of temperature on the reaction activity and stability of phage enzyme. The optimum reaction temperature of phage enzyme was determined by hydrolysis at different temperatures for 0.5 h (–▲–), 2 h (–●–) or 4 h (–■–), respectively. The heat-stability of enzyme was detected by pretreating the crude depolymerase solution at 10℃, 50℃, 60℃ and 70℃ for 0.5 h () or 2 h (), respectively. The residual enzyme activity was monitored according to OD520value by the DNS method. Titer of phage after thermal treatment for 0.5 h () and 2 h () was evaluated by the bilayer agar method.
When the reaction temperature increased from 10℃ to 60℃, the activity of phage enzyme correspondingly increased (Fig.3). The maximum enzyme activity was observed at 60℃ after incubation for 0.5, 2 or 4 h. The enzyme activity began to gradually decrease when the temperature reached 70℃. In addition, it was found that the enzyme had a distinct heat-stability under the experimental conditions. Almost all measurable activity was retained after treated at 60℃ for 30 min, whereas the phage titer on the host bacteria decreased from 6.45×108to 1.6×106pfu mL−1, showing that the phage is heat-sensitive. After pretreatment at 70℃ for 30 min, the phage particles were completely inactivated, whereas the majority of enzyme activity (>90%) remained. The obvious difference in heat-sensitivity between the phage particle and EPS depolymerase is advantageous for isolation and purification of the phage enzyme. The application of heat-stable phage enzyme can effectively inhibit bacterialcontamination during oligosaccharides preparation and exclude the influence of phage on the biofilm. Similar results have been reported in the literatures. For example, a phage depolymerase isolated fromK. pneumoniaeB5055 was notable as it was more heat-stable than the associated phage, and 70% of enzyme activity remained after incubation at 60℃ for 30 min (Kassa and Chhibber, 2012). Sutherland (1967) also demonstrated that the enzyme extracted from the phage F31 showed great degradation activity above 55℃ against the EPS fromK. aerogenes. It is obvious that the phage enzyme isolated in this work possesses substantially higher heat-stability than those reported in previous work.The crude depolymerase showed highest activity at pH 6.5 (Fig.4). The enzyme was partly destroyed when exposed to pH below 5.5 or above 8.0. The cations K+, Na+and Ca2+had promoting effects on the enzyme activity. Among these, 0.1 mol L−1K+and Na+elevated the relative enzyme activity by 46% (Table 2). No enzyme activity of depolymerase was observed in the presence of 0.1 mol L−1EDTA after incubation at 60℃ for 4 h.
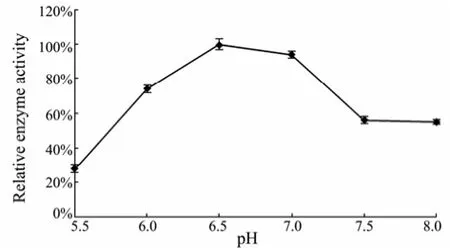
Fig.4 Effects of initial pH values on the phage enzyme activity.
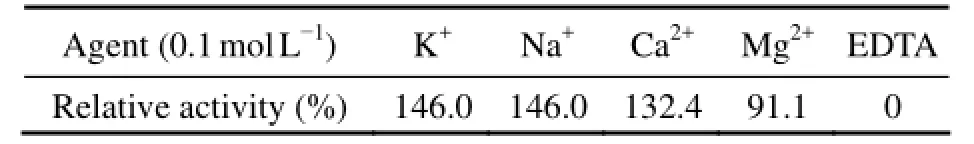
Table 2 Effects of metal cations and EDTA on relative enzyme activity
3.5 Electron Micrographs of Phage P13 Particles and Its Nucleic Acid Analysis
After concentration by PEG 20000, the titer of phage reached 2.6×1010pfu mL−1. Before the extraction of phage nucleic acid, DNA and RNA must be entirely removed from the host cells by nuclease. The phage nucleic acid extract showed a single band on agarose gel, with a molecule larger than 20 kbp (Fig.5). Its nucleic acid was digested by DNase I and insusceptible to RNase A, showing that it was a DNA phage. After treated by restriction endonucleaseNdeI, phage DNA was digested and produced four bands on agarose gel, whereas the nucleic acid fromKlebsiellaK13 was resistant toNdeI. These proved that phage P13 DNA extracted after DNase I and RNase A treatment was highly pure without contamination of host cell DNA. SinceNdeI is a restriction enzyme capable of digesting double-stranded DNA with the restriction cutting site of CA/TATG, the phage was confirmed as a double-stranded DNA virus. High-throughput sequencing of the phage genome showed that the molecular size of its nucleic acid is 45976 bp (data not shown). The size of phage P13 genome detected by agarose gel electrophoresis was far less than its actual size, presumably due to the limiting resolution of routine agarose gel that was unable to separate DNA with a size larger than 20 kbp (Zhu, 2010). TEM showed that the phage P13 consisted of a small spherical head with a diameter of 50 nm and a short stumpy tail spikes (Fig.6), similar to the members ofPodoviridaefamily. The whole DNA sequence of phage P13 was analyzed by BLASTP, showing a high homology with several members of SP6-like virus genus,e.g.,Enterobacteriaphage SP6 (97%), K1E (88%), K1-5 (91%) andErwinia amylovoraphage Era103 (77%). The molecular size of nucleic acid of P13 is much close to that of SP6-like virus genus, which has 44.7 kbp genomes on average (King, 2011). Accordingly, phage P13 is ascribed to the familyPodoviridae.
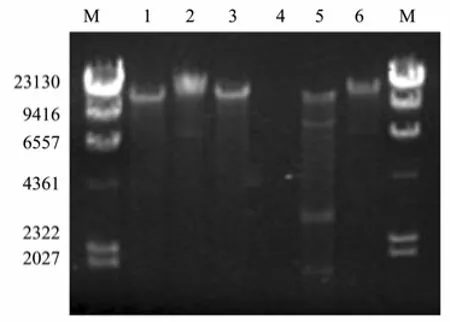
Fig.5 Agarose gel electrophoresis of phage P13 nucleic acid. The electrophoresis was performed in 0.8% agarose gel in 1×TAE buffer with a constant voltage of 95 V. Lane M, molecular size marker (λ-Hind Ⅲ digest DNA Marker); lane 1, phage nucleic acid; lane 2, Klebsiella K13 DNA; lane 3, phage nucleic acid digested by RNase A; lane 4, phage nucleic acid digested by DNase I; lane 5, phage DNA treated by restriction enzyme Nde I; lane 6, Klebsiella K13 DNA treated by Nde I.
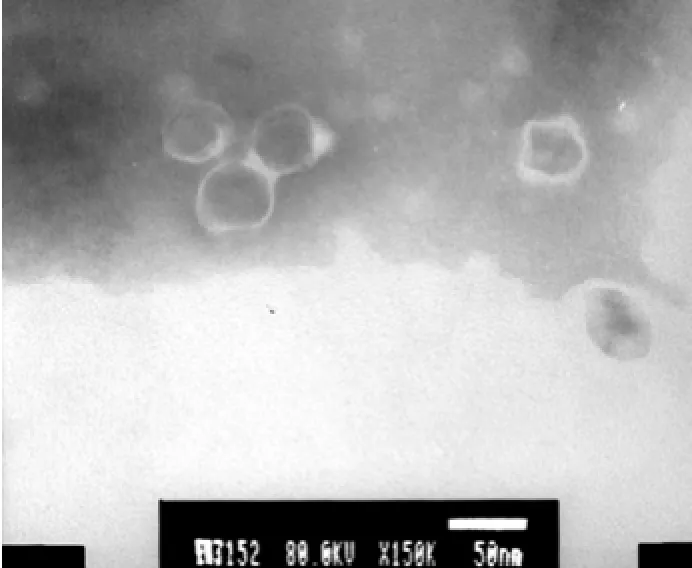
Fig.6 TEM micrograph of phage P13 particles.
4 Conclusions
This work isolated a bacteriophage-borne EPS depolymerase showing high efficiency of bacterial EPS degrada-tion, and proposed the preparation method of special oligosaccharides. Acetone precipitation and ultrafiltration centrifugation are feasible procedures for the preparation of phage P13 enzyme. Thermal treatment at 70℃ for 30 min can be used for the complete separation of phage enzyme from phage particles due to the distinct heat stability of the enzyme. This property can greatly contribute to the application of phage enzyme in production of novel oligosaccharides and bio-control of bacterial biofilm. The isolated phage is a double-stranded DNA virus with a nucleic acid molecule of 45976 bp. The propagation of phage likely depends on the growth of host bacterial cells, which restricts the large-scale preparation of phage enzyme. Therefore, gene cloning of phage depolymerase enzyme and expression in bacterial cells may be the only way for future development of the enzyme.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No.41076087).
Adams, M. H., and Park, B. H., 1956. An enzyme produced by a phage-host cell system Ⅱ. The properties of the polysaccharide depolymerase. Virology, 2: 719-736.
Altmann, F., Christian, R., Czerny, T., Nimmich, W., and Marz, L., 1990. Bacteriophage-associated glycan hydrolases specific for Escherichia coli capsular serotype K12. European Journal of Biochemistry, 189: 307-312.
Bayer, M. E., and Anderson, T. F., 1963. The preparation of holey films for electron microscopy. Cellular and Molecular Life Sciences, 19: 433-434.
Cescutti, P., Toffanin, R., Kvam, B. J., Paoletti, S., and Dutton, G. G., 1993. Structural determination of the capsular polysaccharide produced by Klebsiella pneumoniae serotype K40. NMR studies of the oligosaccharide obtained upon depolymerisation of the polysaccharide with a bacteriophage-associated endoglycanase. European Journal of Biochemistry, 213: 445-453.
Coenye, T., and Nelis, H. J., 2010. In vitro and in vivo model systems to study microbial bio fi lm formation. Jounal of Microbiol Methods, 83: 89-105.
Davies, D., 2003. Understanding bio fi lm resistance to antibacterial agents. Nature Review Drug Discovery, 2: 114-122.
Di Fabio, J. L., Karunaratne, D. N., and Dutton, G. G., 1985. Novel oligosaccharides obtained by bacteriophage degradation of the polysaccharide from Klebsiella serotype K26. Carbohydrate Research, 144: 251-261.
Dutton, G. G. S., Di Fabio, J. L., Leek, D. M., Merrifield, E. H., Nunn, J. R., and Stephen, A. M., 1981. Preparation of oligosaccharides by the action of bacteriophage-borne enzymes on Klebsiella capsular polysaccharides. Carbohydrate Research, 97: 127-138.
Flint, S. H., Bremer, P. J., and Brooks, J. D., 1997. Bio fi lms in dairy manufacturing plant description, current concerns and methods of control. Biofouling, 11: 81-97.
Griffiths, A. J., and Davies, D. B., 1991. Type-specific carbohydrate antigens of pathogenic bacteria. Part 1: Enterobacteriaceae. Carbohydrate Polymers, 14: 241-279.
Habash, M., and Reid, G., 1999. Microbial biofilms: Their development and significance for medical device-related infections. The Journal of Clinical Pharmacology, 39: 887-898.
Hanlon, G. W., Denyer, S. P., Olliff, C. J., and Ibrahim, L. J., 2001. Reduction in exopolysaccharide viscosity as an aid to bacteriophage penetration through Pseudomonas aeruginosa biof i lms. Applied and Environmental Microbiology, 67: 2746-2753.
Hughes, K. A., Sutherland, I. W., and Jones, M. V., 1998. Biof i lm susceptibility to bacteriophage attack: the role of phage-borne polysaccharide depolymerase. Microbiology, 144: 3039-3047.
Kassa, T., and Chhibber, S., 2012. Thermal treatment of the bacteriophage lysate of Klebsiella pneumoniae B5055 as a step for the purification of capsular depolymerase enzyme. Journal of Virological Methods, 179: 135-141.
Kim, W. S., and Geider, K., 2000. Characterization of a viral EPS-depolymerase, a potential tool for control of fire blight. Phytopathology, 90: 1263-1268.
King, A. M. Q., Adams, M. J., Carstens, E. B., and Lefkowitz, E. J., 2011. Virus taxonomy: ninth report of the international committee on taxonomy of viruses. In: Genus: SP6-Like Viruses. Elsevier Academic Press, San Diego, 75-76.
Mou, H., Wang, J., Jiang, X., and Liu, Z., 2008. Preparation and properties of bacteriophage-borne enzyme degrading bacterial exopolysaccharide. High Technology Letters, 14: 210-215.
Roberts, T. S., 1996. The biochemistry and genetics of capsular polysaccharide production in bacteria. Annual Review of Microbiology, 50: 285-315.
Scorpio, A., Tobery, S. A., Ribot, W. J., and Friedlander, A. M., 2008. Treatment of experimental anthrax with recombinant capsule depolymerase. Antimicrobial Agents and Chemotherapy, 52: 1014-1020.
Simoes, M., Simoes, L. C., and Vieira, M. J., 2010. A review of current and emergent biof i lm control strategies. LWT-Food Science and Technology, 43: 573-583.
Stewart, P. S., Murga, R., and Srinivasan, R., 1995. Biofilm structural heterogeneity visualized by three microscopic methods. Water Research, 29: 2006-2009.
Sutherland, I. W., 1967. Phage-induced fucosidase hydrolysing the exopolysaccharide of Klebsiella aerogenes type 54. Biochemical Journal, 104: 278-285.
Sutherland, I. W., 2001. Biofilm exopolysaccharides: A strong and sticky framework. Microbiology, 147: 3-9.
von Borel, E., Hostettler, F., and Deuel, H., 1952. Quantitative zuckerbestimmung mit 3, 5-dinitrosalicylsaure and phenol. Helvetica Chimica Acta, 35: 115-120.
Xu, G., and Chen, F., 2010. Biochemistry and technology practical training. In: Protein Content Detection by Coomassie Brilliant Blue Staining. Huazhong University of Science and Technology Press, Wuhan, 43-45.
Yurewicz, E. C., Ghalambor, M. A., Duckworth, D. H., and Heath, E. C., 1971. Catalytic and molecular properties of a phage-induced capsular polysaccharide depolymerse. Journal of Biological Chemistry, 246: 5607-5616.
Zhu, X., 2010. The experimental guide for gene engineering. In: Nucleic Acid Electrophoresis. Higner Education Press, Beijing, 74-77, 227-235.
(Edited by Qiu Yantao)
* Corresponding author. Tel: 0086-532-82032290
E-mail: mousun@ouc.edu.cn
(Received May 12, 2012; revised July 5, 2012; accepted November 19, 2012)
© Ocean University of China, Science Press and Springer-Verlag Berlin Heidelberg 2014
杂志排行

Journal of Ocean University of China的其它文章
- Comparative Study on the Allergenicity of Different Litopenaeus vannamei Extract Solutions
- Isolation and Characterization of a Fucoidan-Degrading Bacterium from Laminaria japonica
- Toxicity of Five Phenolic Compounds to Brine Shrimp Artemia sinica (Crustacea: Artemiidae)
- Application of CFD Modeling to Hydrodynamics of CycloBio Fluidized Sand Bed in Recirculating Aquaculture Systems
- DNA Barcoding Assessment of Green Macroalgae in Coastal Zone Around Qingdao, China
- QSAR for Photodegradation Activity of Polycyclic Aromatic Hydrocarbons in Aqueous Systems