电压门控钠离子通道与疼痛症
2014-04-29韩鹏刚范崇旭陈冀胜
韩鹏刚 范崇旭 陈冀胜
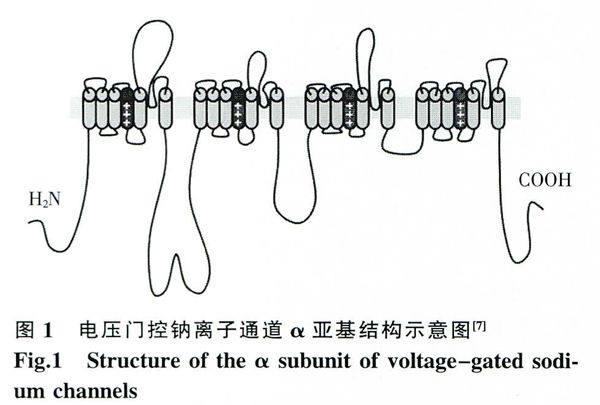
摘要:疼痛是一种常见的疾病和临床症状,有时会严重影响患者的生活质量,因此,疼痛的研究、治疗具有重要的实际意义。电压门控钠离子通道在神经元动作电位的起始和传导中起着关键作用,尤其是亚型Nav1.3、Nav1.7、Nav1.8和Nav1.9,它们广泛存在于背根神经节中,参与了疼痛的形成。其中,Nav1.7的基因突变会导致多种遗传性疾病。因此,这些亚型是潜在的、理想的疼痛治疗靶点。主要对电压门控钠离子通道与疼痛有关的最新研究进展进行了综述。
关键词:电压门控钠离子通道;背根神经节;神经性疼痛;炎症痛
中图分类号:Q955
文献标识码:A
文章编号:1007-7847(2014)03-0275-08
疼痛是一类复杂的感觉,国际疼痛研究协会(International Association for the Study of Pain,ISAP)将疼痛定义为,“一种不愉快的感觉和情绪体验,往往伴随有实际的或潜在的组织伤害”[1]。疼痛可以作为一种警戒信号,提醒机体注意潜在的危险,对机体正常的生命活动具有不可或缺的保护作用。同时,疼痛也是一种常见的临床症状,在引发疼痛的外界刺激消失后,强烈或持久的疼痛会造成生理功能的紊乱,严重影响生命体的生活质量。据报道,全世界约五分之一的人受到中度或严重慢性疼痛的困扰[1]。因此,对于疼痛的研究、治疗药物的开发具有重要的实际意义。
疼痛起源于周围神经系统的伤害感受器。这是一种游离的神经末梢,广泛分布于全身的皮肤、肌肉、关节和内脏组织中,它可以将感受到的热的、机械的或化学的刺激转化为神经冲动(动作电位)并经由传入神经纤维传递到其位于背根神经节(dorsal root ganglia,DRG)的胞体部分[2],最终传递到高级神经中枢,引起痛觉。而神经元中动作电位的产生和传导又依赖于细胞膜上的电压门控钠离子通道(voltage-gated sodium channels, VGSCS)。当细胞膜去极化时,钠离子通道激活,通道打开,引起钠离子内流,使细胞膜进一步去极化,导致动作电位的产生。因此,抑制异常的钠离子通道活动有助于疼痛的治疗、缓解。
1电压门控钠离子通道
1.1钠离子通道的组成
电压门控钠离子通道广泛存在于可兴奋细胞如神经元、骨骼肌细胞的细胞膜上,是一类跨膜糖蛋白复合体,由一个α亚基和数个β亚基构成[3]。
α亚基是钠离子通道的功能载体,由1700~2000个氨基酸组成[4],这些氨基酸形成了4个结构域(Domain,I-Ⅳ),每个Domain又包含6个跨膜片段(S1~S6)(图1)。Domain之间由一些大的胞内环连接起来,而各片段之间则由一些小的胞外或胞内环相连接。其中,S4上含有丰富的碱性氨基酸残基,被认为是钠离子通道的电压敏感元件。当细胞膜去极化时,S4上的正电荷可以沿着S4的轴线做顺时针旋转往外迁移,改变钠离子通道构象,使通道打开。S5和S6之间的发夹环(pore loop,P-loop)形成了微孔外部,与对钠离子的选择性有关,微孔的胞内部分则由S6围成。连接DomainⅢ和Ⅳ的胞内环起着失活阀门的作用,它可以折叠进入微孔的胞内开口,阻塞微孔,使钠离子通道失活[3]。
β亚基主要起辅助作用,可以对离子通道的动力学以及电压门控依赖性进行修饰,目前已经鉴定了4种亚型(β1~β4)[5]。β亚基由胞外的免疫球样蛋白折叠、一个简单的跨膜片段和胞内C端组成,通过共价键或疏水相互作用与α亚基相连。有研究表明,β亚基参与了钠离子通道在一些特殊位置的定位,以及与黏附分子、细胞外基质和细胞骨架的相互作用[6]。
1.2钠离子通道亚型
根据α亚基的不同可以对钠离子通道进行分类,目前,已经在哺乳动物中鉴定了9种钠离子通道亚型[8],由于其氨基酸序列相似度均大于50%,因此被认为来自同一家族,即Navl(Navl.l~Nav1.9)。不同亚型表现出不同的组织分布和电生理、药理学特征[9,10]。根据能否被纳摩尔河豚毒素(tetro -dotoxin,TTX)有效抑制,钠离子通道被分为TTX敏感型(TTX-sensitive,TTX-S)和TTX不敏感型(TTX-resistant,TTX-R)[9]。其中,Nav1.1、Nav1.2、Nav1.3和Nav1.7为TTX-S型,编码基因位于人类染色体2q23 -24,它们在神经元中大量表达。Nav1.5、Nav1.8和Nav1.9为TTX-R型,编码基因位于人类染色体3p21-24[11]。其中,Nav1.5主要存在于心肌细胞中,Nav1.8、Navl.9存茌子同围神经系统(peripheral nervous system,PNS)。另外两种亚型,Nav1.4和Nav1.6都为TTX-S型,分别在骨骼肌和中枢神经系统(central nervous system,CNS)中大量存在[l2]。与疼痛相关的亚型主要是Nav1.3、Nav1.7、Nav1.8和Nav1.9[1],下文将重点讨论。
2DRG中的钠离子通道亚型
作为痛觉传人的第一级神经元,DRG神经元在痛觉的形成中发挥着重要作用。与其他类型的神经元相比,DRG神经元中表达了多种钠离子通道,包括亚型Nav1.1、Nav1.6、Nav1.7、Nav1.8和Nav1.9[13]。
Nav1.3是胚胎时期神经元中最主要的钠离子通道亚型,但随着个体发育其表达不断下降,在成熟的啮齿动物DRG中已经无法检测到[l4],仅少量存在于大脑中[15]。有研究表明,Nav1.3的一个突变与儿童期癫痫有关[16],这与刚出生时人大脑中有着高水平的Nav1.3的事实是相关的[17]。目前,只在哺乳动物中发现了Nav1.7、Nav1.8和Nav1.9,这说明它们在进化上出现较晚。此外,Nav1.7、Nav1.8和Nav1.9只存在于PNS,并在各种伤害感受神经元中广泛分布,这也许表明它们在包括疼痛在内的感觉形成中有着重要作用[18~20]。
3与疼痛有关的钠离子通道亚型
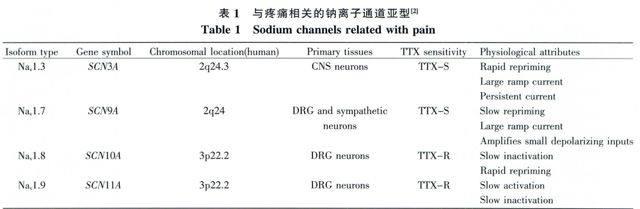
根据疼痛产生的病理生理学特征,可以将疼痛分为炎症痛(inflammatory pain)和神经性疼痛(neuropathic pain)。炎症痛是指组织损伤产生的各种介质如前列腺素、腺苷等刺激感觉神经末梢引起的疼痛,而神经性疼痛中一般没有组织损伤,往往是由于中枢神经系统或周围神经系统损伤或损伤后功能紊乱所引起的疼痛。目前,普遍认为与疼痛相关的钠离子通道包括Nav1.3、Nav.7、Nav1.8 和Nav1.9(表1),它们在炎症痛和神经性疼痛的形成中有着不同的作用。
3.1Nav1.3
Nav1.3为TTX-S型,编码基因为SCN3A,在成体PNS中含量很低,仅少量存在于交感神经元[21],其生理特点为失活后迅速恢复,可以产生持续电流。在弱去极化条件下,Nav1.3会形成慢速失活,产生斜坡电流,起去极化放大作用[22]。
实验表明,当外周神经受损时,Nav1.3在DRG、背侧角神经元中的表达都会上升[23,24]。脊神经损伤实验里,Nav1.3在背侧角神经元中的表达也会上升[25]。此外,三叉神经性疼痛患者牙龈组织中Nav1.3的含量也明显提高[26]。在坐骨神经和脊神经损伤试验中,Nav1.3的反义寡核苷酸能降低Nav1.3在背侧角神经元中的表达,同时疼痛也得到明显缓解[27]。以上这些实验似乎都表明Nav1.3与疼痛密切相关,但是,另一个表达下调(knock-down)实验中,Nav1.3的反义寡核苷酸可以降低Nav1.3的表达水平,却不能缓解由小鼠外周神经损伤引起的疼痛[24]。而且,大鼠的神经损伤模型中,全部敲除(konck-out)Nav1.3或选择性敲除DRG中的Nav1.3都不能缓解疼痛[28],这存在两种解释:一是Nav1.3与受损神经元的过度兴奋无关,二是存在一种补偿机制替代了Nav1.3的缺失。因此,Nav1.3与疼痛存在一定的联系,但具体的作用尚不清楚,有待进一步研究。
3.2Nav1.7
Nav1.7为TTX-S型,编码基因为SCN9A,主要存在于DRG神经元、交感神经元[20,29]、施万细胞、神经内分泌细胞以及嗅上皮中[30],其电生理特点为快速激活、迅速失活及缓慢恢复,弱去极化条件下发生慢速失活,可以产生大的斜坡电流[31]。因此,Nav1.7被认为可以放大神经元中的启动电位,是动作电位的开端[10]。
在大鼠DRG轴突切断实验中,受损DRG神经元中Nav1.7 mRNA的表达水平明显下降[32],同时观察到TTX-S电流由缓慢恢复转化为快速恢复。与动物实验结果一致,人的外周神经损伤后,DRG神经元中Nav1.7的表达水平也有所下降[33,34]。这些证据似乎都说明Nav1.7参与了神经性疼痛的形成,但是敲除Nav1.7不会影响正常的神经性疼痛[35]。因此,Nav1.7在神经性疼痛中的作用还有待于进一步研究确认。
动物实验模型表明,炎症会引发DRG神经元中Nav1.7的表达上升[23,36]表达下调实验中,Nav1.7表达下调可以明显缓解由弗氏完全佐剂(completefreund's adjuvant,CFA)引起的小鼠热痛觉过敏症状(thermal hyperalgesia),这也说明Nav1.7参与了炎症痛的形成[37]。在同时表达Nav1.7、Nav1.8的小鼠DRG神经元中选择性地敲除Nav1.7,结果炎症痛得到明显改善[38],这进一步说明了Nav1.7在炎症痛中扮演着重要角色。 此外,SCN9A突变会引起多种疼痛类遗传疾病,这也说明在Nav1.7在痛觉形成中有着重要作用,下文会详细介绍。
3.3Nav1.8
Nav1.8为TTX-R型,编码基因为SCN10A,主要存在于三叉神经节神经元和DRG神经元中,具有慢速失活、迅速恢复的电生理特征[19]。在表达Nav1.8的神经元内,动作电位的上升支主要由Nav1.8电流构成[39]。
目前没有充分证据表明Nav1.8参与了神经性疼痛的形成[40]。坐骨神经轴突横切模型中,Nav1.8在受损神经元中的mRNA和蛋白水平都明显降低[41.42]。然而,在研究神经性疼痛的一些模型中,神经损伤会使Nav1.8在轴突和神经元胞体中的表达水平上升[43,44]。在小鼠慢性缩窄性损伤(chro-nic constriction injury,CCI)模型中,使用Nav1.8反义寡核苷酸在降低Nav1.8表达的同时可以明显地缓解疼痛[45],用siRNA也有同样的作用[46]。A-803467是Nav1.8的选择性抑制剂,它可以缓解神经性疼痛症状[47],其他一些毒素抑制实验也有相似效果[48,49],这些似乎都表明Nav1.8在神经性疼痛中发挥了一定的作用。但是,敲除Nav1.8却不影响正常的神经性疼痛行为[50]。此外,同时敲除Nav1.7、Nav1.8也不能缓解神经性疼痛[35]。
已经证实Nav1.8在炎症痛中发挥了一定的作用。大鼠爪内注射角又菜胶(carrageenan)后,DRG神经元中Nav1.8的表达有所上升[23]。表达下调实验中,siRNA使Nav1.8表达下调后炎症痛得到明显缓解[45]。Nav1.8的选择性抑制剂A-803467也能缓解炎症痛[47]。敲除实验则进一步确认了它在炎症痛中的作用,Nav1.8敲除小鼠不能表现出正常的内脏炎症痛[51,52]。
总之,Nav1.8在神经性疼痛中的作用还有待于进一步研究,而在炎症痛的作用已经得到确认。
3.4Nav1.9
Nav1.9为TTX-R型,编码基因为SCN11A,主要分布在感受伤害的DRG神经元[53,54]、三叉神经节和肠肌层神经元[54,55]。Nav1.9的激活电压接近神经元的静息膜电位(-60~-70mV),具有缓慢激活、缓慢失活的动力学特征,因此可以产生持续较久的TTX-R电流[56],这表明Nav1.9没有参与动作电位上升支的形成,但它可以放大和延长神经元对阈下去极化的反应,引发动作电位[57,58]。在人体内,Nav1.9的激活电压为-80mV,这比啮齿动物DRG神经元中Nav1.9的激活电压低10~20mV,这可能是由于不同物种中Nav1.9的氨基酸序列存在一定差异[59]。
轴突切断后,Nav1.9在受损DRG神经元的mRNA、蛋白和电流水平都有所下降[53]。但是,在小鼠的慢性疼痛模型中,用Nav1.9的反义寡核苷酸处理能降低Nav1.9的表达,但不能缓解神经性疼痛症状[60]。敲除Nav1.9对神经性疼痛也没有影响[61]。不过,有研究发现,Nav1.9的mRNA水平的下降程度与疼痛严重程度成反比:在疼痛最严重的动物中下降得最少,最小疼痛的动物中下降得最多[62],这表明Nav1.9也许对疼痛阈值有影响。
Nav1.9在炎症痛中起着一定作用。在大鼠炎症痛模型中,Nav1.9在DRG神经元中的表达明显增加[63]。敲除Nav1.9会破坏身体炎症痛机制[61,64]。但即使是在炎症痛中,它的作用也是不明确的,因为采用反义寡脱氧核苷酸使Nav1.9的表达下降,却不能减轻由完全弗氏佐剂诱导产生的热超敏症[65]。
综上所述,有证据表明Nav1.9参与了炎症痛的形成,但在神经性疼痛中的作用还有待于进一步研究。
4与钠离子通道突变有关的疼痛疾病
钠离子通道的编码基因发生突变会改变钠离子通道的结构,影响其正常功能的发挥,导致遗传性疾病的发生,这些疾病包括癫痫、周期性麻痹、心肌传导病等。其中,与疼痛相关的突变主要是编码Nav1.7的SCN9A产生,分为功能获得性突变和功能缺失性突变。SCN9A的功能获得性突变主要与遗传性红斑肢痛病(inherited erythromelalgia,IEM)和阵发性极度疼痛障碍(paroxysmal extremepain disorder,PEPD)有关,而功能缺失性突变主要与先天性痛觉缺失(congenital insensitivity topain,CIP)有关。
4.1遗传性红斑肢痛病(inherited erythromelal-gia, IEM)
红斑肢痛病是一种极其罕见的疾病,最早发现于1878年,通常在童年或青春期首次发病,发作时四肢浮肿、极度疼痛,并伴有局部皮肤温度上升和红斑[39,66],往往由体育锻炼和热刺激诱发,通过对患肢进行冷敷可以缓解症状[66,67]。红斑肢痛病分为原发性和继发性,继发性红斑肢痛病与一些血液疾病(如血小板增多病、红细胞增多症)、自身免疫疾病等相关,而越来越多的证据表明原发性红斑肢痛病与SCN9A突变有关。
2004年,Yang等在中国一个IEM患病家族和一个散发病例中发现了两个SCN9A突变[68]。这两个突变分别位于DomainⅡ的S5(突变L858H)以及DomainⅡ中S4、S5之间的连接环(突变1848T)。在许多钠离子通道中,这两个位置的亮氨酸和异亮氨酸都十分保守。这些突变被认为造成了感觉神经元和交感神经元的过度兴奋,导致患者的神经性疼痛和血管收缩。之后,又发现了一个患病的美国家族,这个家族几代人中共有36名IEM患者,对其中17人进行深入研究。结果在这17名患者中都发现了一个氨基酸替换突变(F1449V)[69]。随后,在IEM患者中陆续发现了新的Nav1.7突变[70]。Lee等在两例散发IEM患者中发现了突变V1316A,并利用膜片钳研究了温度埘通道电生理的影响,结果表明,较高温度会增强离子通道的活性,这也许是IEM往往由热刺激引发的原因[71]。
目前,已经发现19个与IEM相关的SCN9A突变,包含18个错义突变和1个框内缺失突变[71]。这些突变会改变钠离子通道的电生理特征,使得去激活(deactivation)速度变慢,增加斜坡电流,并朝着超极化的方向改变通道的电压依赖性。在细胞水平,IEM突变会降低DRG神经元产生单个动作电位所需的阈值下限,并增加活化速率[26],使得神经元的兴奋性增强。
目前,临床上还缺乏IEM的有效治疗方法,运用钠离子通道抑制剂治疗IEM具有很大的潜力。分别利用抑制剂卡马西平(Carbamazepine)和利多卡因(Lidocaine)治疗含突变V872G[72]和V400M的IEM患者[73],都取得了不错的疗效。但也有人用美西律(Mexiletine)、利多卡因等钠离子通道抑制剂治疗IEM却没有得到理想的结果[71,74,75]。这可能与不同的突变有关,一些突变会改变钠离子通道的结构,导致其与抑制剂的结合力下降[76]。
4.2阵发性极度疼痛障碍(paroxysmal extremepain disorder,PEPD)
PEPD是由SCN9A突变引起的另一种常染色体显性遗传疾病,最早发现于1959年[38]。PEPD通常在患者年龄很小的时候就发作,有的在婴儿期甚至还在子宫中时就开始,典型症状为突发性烧灼痛,并伴有潮红等症状。PEPD往往由于肠运动和小:周探测触发,并伴随有晕厥、心动过缓、心搏停止等[39,77,78]。在发作初期,PEPD只会影响直肠区域,但随着年龄的发展症状会逐渐扩展到眼部、上颌、下颌,并由温度改变、饮食、低落的情绪等诱发,一般持续几分钟,严重时可持续数小时。
2006年,Fertleman[78]等首先在PEPD患者中发现、鉴定了Nav1.7突变。他们在l1个家族性患者和两个散发病例中鉴定了8个SCN9A错义突变,并对其中3个突变(11461T、T14641、M1627K)进行了分析,结果表明突变通道的失活过程被破坏,产生了长达几百毫秒的持续电流。相比于野生型通道,M1627K突变通道具有更快的失活恢复速率[79]。电生理分析表明PEPD突变与IEM突变不同,PEPD是朝着去极化的方向改变失活的电压依赖性,增加斜坡电流,减慢失活;而IEM是朝着超极化的方向改变钠通道激活的电压依赖性[2,80]。由此可见,改变失活的稳定性对神经元的兴奋性有着深远的影响。此外,研究表明,PEPD突变会产生更强的、由β4亚基介导的复苏电流[81]。
虽然IEM和PEPD突变都会增强Nav1.7的活性,但是却有着不同的临床症状,这需要进一步研究。钠离子通道抑制剂对PEPD有治疗作用,如用抗惊厥药物卡马西平治疗PEPD就有着不错的疗效[79]。
4.3IEM-PEPD混合症状(mixed IEM - PEPDphenotype)
如上所述,两类不同的SCN9A突变分别引起了两种具有不同症状的、完全独立的疾病:IEM和PEPD。但是,2008年,Estacion等在一个特殊的患者中鉴定了一个新的Nav1.7突变:AI632E,该患者同时表现出IEM和PEPD的典型症状[82]。A1632E突变改变了DomainⅣ上S4、S5连接环中一个高度保守的氨基酸。除了Nav1.9,所有钠离子通道亚型在这个位置都是丙氨酸。该名患者自出生就表现出呼吸暂停、心动过缓和食欲低下。随着年龄增加,患者表现出对直肠部位敏感、疼痛和红斑症状。同时,四肢也表现出疼痛和红斑,并往往由热刺激或触摸引发,冷敷可以明显地缓解症状。
电生理分析表明A1632E突变通道同时具有PEPD和IEM突变通道的动力学特征。A1632F突变朝着去极化的方向减慢快速失活,这导致了持续电流,同时朝着超极化的方向改变激活的电压依赖性,减慢失活,加速恢复,增加对微弱的斜坡型刺激的去极化反应。分析还表明A1632E突变亦能增加DRG神经元和三叉神经节神经元对阈上刺激的活化频率。
鉴于A1632E突变可以同时表现出PEPD和IEM症状,因此可以看做是两种疾病之间的一种联系。在此基础上,有人提出这两种疾病有可能是来自同一个致病机制[82],是一个连续生理过程的不同阶段。
4.4先天性痛觉缺失(congenital insensitivity topain, CIP)
SCN9A的功能缺失性突变会引起一种常染色体隐性遗传疾病,即先天性痛觉缺失(CIP),这是一种极其罕见的疾病,直到20世纪初才报道了第一例。2006年,Cox等[83]对3个有血缘关系的巴基斯坦家庭中的CIP患者进行了研究,这些患者从来没有在任何时间、身体任何部位感觉到疼痛,但其他一切正常。Cox首次在这些患者中鉴定了Nav1.7的隐形纯合子无义突变(S459X、1767X和W897X),证实该病为常染色体隐性遗传。经分析,这些突变导致了截短通道蛋白的合成,使得感受伤害的神经元中Nav1.7失去正常生理功能。为了证实这点,他们在HEK293细胞中分别表达野生型和CIP突变型Nav1.7,然后进行电压钳分析,结果发现突变型通道不能产生正常的电流。
后来,陆续又报道了一些CIP患者或家庭的研究情况,其中都发现了Nav1.7的功能缺失性突变[84,85]。Hong等在来自7个不同民族的9个CIP患者中鉴定了10个Nav1.7突变[85]。有的家庭中,杂合子的父母拥有正常的痛觉,这说明一个等位基因的丢失不会造成单倍剂量不足。除了缺失痛觉,CIP患者在神经病学检查中一切正常,其他身体检查也没有显示任何异常,这说明CIP完全是南于钠离子通道功能丧失引起,这也证实Nav1.7在疼痛的形成中有着重要作用。
除了引发以上几种疾病外,最近还发现Nav1.7突变与慢性疼痛综合症(chronic pain syn-drome)和小纤维神经病(small fiber neuropathy,SFN)有关。2011年,Dabby等[86]首次在原发性严重持续性神经性疼痛(idiopathic severe continuousneuropathic pain)患者中鉴定了新的Nav1.7功能获得性突变( W1550R)。该突变会改变DomainⅣ中S2的一个保守氨基酸(色氨酸),增加蛋白疏水性。Farber等[87]在28个自发性SFN患者中的8名患者中鉴定了新的Nav1.7突变。电压钳分析表明这些突变都是功能获得性的,但是突变通道的电生理特征不同于IEM和PEPD突变通道。这些新突变的发现表明钠离子通道亚型广泛参与了多种疼痛疾病的形成。
5展望
鉴于钠离子通道在疼痛形成中的重要角色,且Nav1.7、Nav1.8和Nav1.9只存在于周围神经系统内,以它们为靶点治疗疼痛时,理论上可以尽量把药理作用限制在周围神经系统范围内,减少副作用的发生,因此是良好的疼痛治疗靶点。
临床上,钠离子通道抑制剂应用较早,如卡马西平、利多卡因、美西律等用于治疗疼痛,但是它们对钠离子通道亚型缺乏足够的选择性,因而产生心脏毒性和中枢神经副作用[88,89],这极大地限制了其运用。因此,寻找具有亚型选择性、高亲和力的钠离子通道抑制剂作为与疼痛有关疾病的治疗药物,将是很长一段时间内的研究重点。天然毒素是钠离子通道选择性抑制剂的主要来源,如芋螺毒素(Conotoxins)、蜘蛛毒素(Spider toxins)、蝎毒素(Scorpions)等,它们通过与通道上的毒素受体位点相互作用而调节钠离子通道功能,如芋螺毒素μo-MrⅥB可以选择性地作用于Navl.8,表现出镇痛活性[49]; μ-KⅢA主要作用于TTX-R亚型,也表现出一定的镇痛活性。在天然毒素中筛选或设计改造出具有更高亚型选择性的多肽化合物已经成为了研究热点,这也许代表了疼痛治疗的新希望。
参考文献( References):
[1]THEILE J W,CUMMINS T R.Recent developme,nts regardingvoltage-gated sodium channel blockers for the treatment of in—herited and acquire,d neuropathic pain syndromes[J]. Frontiersin Pharmacology, 2011,2:54.
[2]DIB-HAJJ S D,CUMMINS T R.BLACK J A,et al.Sodiumchannels in normal and pathological pain[J]. Annual Review ofNeuroscience, 2010,33:325-347.
[3]CATTERALL W A.From ionic currents to molecular mecha-nisms: the structure and function of voltage-gated sodium chan-nels[J]. Neuron, 2000, 26(1):13-25.
[4]DUCLOHIER H.Structure-function studies on the voltage-gated sodium channel[J]. Biochimica et Biophysica Acta, 2009,1788(11):2374-2379.
[5]TSENG T T,MCMAHON A M,JOHNSON V T,et al.Sodiummchannel auxiliary subunits[J]. Journal of Molecular Microbiolo-gy and Biotechnology, 2007, 12(3-4):249-262.
[6]BRACKENBURY W J,ISOM L L Na channel beta subunits:Overachievers of the ion channel family[J].Frontiers in Phar-macology, 2011,2:53.
[7]CESTELE S,CATTERALL W A.Molecular mechanisms ofneurotoxin action on voltage-gated sodium channels[J]. Bioch-imie, 2000, 82(9-10):883-892.
[8]GOLDIN A L.Evolution of vollage-gated Na(+)channels[J].The Journal of Experimental Biology, 2002, 205(5):575-584.
[9]CATTERALL W A,GOLDIN A L,WAXMAN S G.Interna-tional Union of Pharmacology. XLVII. Nomenclature and struc-ture-function relationships of voltage-gated sodium channels[J]. Pharmacological Reviews, 2005, 57(4):397-409.
[10]RUSH A M,CUMMINS T R,WAXMAN S G.Multiple sodiumchannels and their roles in electrogenesis within dorsal rootganglion neurons[J], The Journal of Physiology, 2007, 579(1):1-14.
[11]GOLDIN A L Resurgence of sodium channel research[J]. An-nual Review of Physiology, 2001,63:871-894.
[12]FOZZARD H A,HANCK D A.Structure and function of volt-age-dependent sodium channels: comparison of brain II andcardiac isoforms[J]. Physiological Reviews, 1996, 76(3):887-926.
[13]BLACK J A,DIB-HAJJ S,MCNABOLA K,et al.Spinal sen-sory neurons express multiple sodium channel alpha-suhunitmRNAs[J]. Brain Research Molecular Brain Research, 1996. 43(1—2):117-131.
[14]DIB-HAJJ S D,BLACK J A,WAXMAN S G Voltage-gatedsodium channels: therapeutic targets for pain[J]. Pain Medicine,2009, 10(7):1260-1269.
[15]BECKH S,NODA M,LUBBERT H,et al.Differential regula-tion of three sodium channel messenger RNAs in the rat cen-tral nervous system during development[J]. The EMBO Journal,1989, 8(12):3611-3616.
[16]HOLLAND K D.KEARNEY J A,GLAUSER T A,et al.Mu-tation of sodium channel SCN3A in a patient with cryptogenicpediatric partial epilepsy[J]. Neuroscience Letters, 2008,433(1):65-70.[17]WHITAKER W R, FAULL R L WALDVOGEL H J, et al.Comparative distribution of voltage-gated sodium channel pro-teins in human brain[J]. Brain Research. Molecular Brain Re-search, 2001, 88(1-2):37-53.
[18]FANG X, DJOUHRI L, BLACK J A, et al. The presence androle of the tetrodoloxin -resistant sodium channel Na(v)1.9(NaN) in nociceptive primary afferent neurons[J]. The Journal ofNeuroscience: the Official Journal of the Sociely for Neuro-science,2002, 22(17):7425-7433.
[19]DJOUHRl L FANG X, OKUSE K, et al. The TTX-resistantsodium channel Navl.8 (SNS/PN3): expression and correlationwith membrane properties in rat nociceptive primary afferentneurons[J]. The Journal of Physiology, 2003, 550(3):739-752.
[20]DJOUHRI L, NEWTON R, LEVINSON S R, et al. Sensoryand electrophysiological properties of guinea-pig sensory neu-rones expressing Navl.7 (PNI) Na+ channel alpha subunitprotein[J]. The Journal of Physiology, 2003, 546(2):565-576.
[21]WAXMAN S G, KOCSIS J D, BLACK J A. Type III sodiumchannel mRNA is expressed in embryonic but not adult spinalsensory neurons, and is reexpressed following axotomy[J]. Journalof Neurophysiology, 1994, 72(1):466-470.
[22]HERZOG R I, CUMMINS T R, WAXMAN S C. PersistentTTX-resistant Na+ current affects resting potential and re-sponse to depolarization in simulated spinal sensory neurons[J].Journal of Neurophysiology, 2001, 86(3):1351-1364.
[23]BLACK J A, LIU S, TANAKA M, et al. Changes in the ex-pression tetrodotoxin-sensitive sodium channels within dor-sal root ganglia neurons in inflammatory pain[J]. Pain, 2004,108(3):237-247.
[24]LINDIA J A, KOHLER M G, MARTIN W J, et al. Relation-ship between sodium channel Navl.3 expression and neuro-pathic pain behavior in rats[J]. Pain, 2005, 117(1-2):145-153.
[25]LAMPERT A. HAINS B C, WAXMAN S G. Upregulation ofpersistent and ramp sodium current in dorsal horn neurons af-ter spinal cord injury[J].Experimental Brain Research. Experi-mentelle Hirnforschung. Experimentation Cerebrale, 2006, 174(4):660-666.
[26]SIQUEIRA S R, ALVES B, MALPARTIDA H M, et al. Ab-normal expression of voltage-gated sodium channels Nav1.7,Nav1.3 and Nav1.8 in trigeminal neuralgia[J]. Neuroscience, 2009,164(2):573-577.
[27]HAINS B C, SAAB C Y, KLEIN J P, et al. Altered sodiumchannel expression in second-order spinal sensory neuronscontributes to pain after peripheral nerve injury[J]. The Journalof Neuroscierice: the Official Journal of the Society for Neuro-science,2004, 24(20):4832-4839.
[28]NASSAR M A, BAKER M D, LEVATO A, et al. Nerve injuryinduces robust allodynia and ectopic discharges in Navl.3 nullmutant mice[J]. Molecular Pain, 2006,2:33.
[29]SANGAMESWARAN L, FISH L M, KOCH B D, et al. A nov-el tetrodotoxin-sensitive, voltage-gated sodium channel ex-pressed in rat and human dorsal root ganglia[J]. The Journal ofBiological Chemistry,1997, 272(23):14805-14809.
[30]WEISS J, PYRSKI M, JACOBI E, et al. Loss-of-function mu-tations in sodium channel Nav1.7 cause anosmia[J]. Nature, 2011,472(7342):186-190.
[31]HERZOG R I,CUMMINS T R, GHASSEMI F, et al. Distinctrepriming and closed-state inaclivation kinetics of Nav1.6 andNavl.7 sodium channels in mouse spinal sensory neurons[J].The Journal of Physiology, 2003, 551(3):741-750.
[32]CUMMINS T R, WAXMAN S G. Downregulation of tetrodotox-in-resistant sodium currents and upregulation of a rapidlyrepriming tetrodotoxin-sensitive sodium current in small spinalsensory neurons after nerve injury [J].The Journal of Neuro-science: the Official Journal of the Society for Neuroscience,1997, 17(10):3503-3514.
[33]COWARD K, PL,UMPTON C, FACER P, et al. Immunolocal-ization of SNS/PN3 and NaN/SNS2 sodium channels in humanpain states[J]. Pain, 2000, 85(1-2):41-50.
[34]COWARD K, AITKEN A, POWELL A, et al. Plasticity ofTTX-sensitive sodium channels PNl and brain III in injuredhuman nerves[J]. Neuroreport, 2001, 12(3):495-500.
[35]NASSAR M A, LEVATO A, STIRLING L C, et al. Neuropath-ic pain levelops normally in mice lacking both Na(v)1.7 andNa(v)1.8[J]. Molecular Pain, 2005, 1:24.
[36]STRICKIJAND I T, MARTINDALE J C, WOODHAMS P L, etal. Changes in the expression of Navl.7, Navl.8 and Navl.9 ina distinct population of dorsal root ganglia inriervating the ralknee joint in a model of chronic inflammatory joint pain[J].European Journal of Pain, 2008, 12(5):564-572.
[37]YEOMANS D C, LEVINSON S R, PETERS M C, et al. De-crease in inflammatory hyperalge,sia by herpes vector-mediatedknockdown of Navl.7 sodium channels in primary afferents[J].Human Gene Therapy, 2005, 16(2):271-277.
[38]NASSAR M A, STIRLING L C, FORLANI G, et, al. Nocicep-tor-specific gene deletion reveals a major role for Navl.7(PNl) in acute and inflammatory pain[J]. Proceedings of the Na-tional Academy of Sciences of the United States of America,2004, 101(34):12706-12711.
[39]FISCHER T Z, WAXMAN S G. Familial pain syndromes frommutations of the Navl.7 sodium channel[J]. Annals of the NewYork Academy of Sciences, 2010,1184:196-207.
[40]LEO S, D'HOOGE R, MEERT T. Exploring the role of noci-ceptor-specific sodium channels in pain transmission usingNavl.8 and Navl.9 knockout mice[J]. Behavioural Brain Research,2010, 208(1):149-157.
[41]SLEEPER A A, CUMMINS T R, DIB-HAJJ S D, et al.Changes in expression of two tetrodotoxin-resistant sodiumchannels and their currents in dorsal root ganglion neurons af-ter sciatic nerve injury but not thizotomy[J].The Journal of Neu-roscience: the Official Journal of the Society for Neuroscience,2000, 20(19):7279-7289.
[42]DECOSTERD I, JI R R, ABDI S, et al. The pattem of expres-sion of the voltage-gated sodium channels Na(v)1.8 and Na(v)1.9 does not change in uninjured primary sensory neurons inexperimental neuropathic pain models[J].Pain, 2002, 96(3):269-277.
[43]GOLD M S, WEINREICH D, KIM C S, et al. Redistribution ofNa(V)1.8 in uninjured axons enables neuropathic pain[J]. TheJournal of Neuroscience: the, Official Journal of the Society forNeuroscience, 2003, 23(1):158-166.
[44]ZHANG X F, ZHU C Z, THIMMAPAYA R, et al. Differentialaction potentials and firing patterns in injured and uninjuredsmall dorsal root ganglion neurons afler nerve injury[J]. Brain Re-search, 2004, 1009(1-2):147-158.
[45]JOSHI S K, MIKUSA J P, HERNANDEZ G, et al. Involvementof the TTX-resistant sodium channe; Navl.8 in inflammatoryand neuropathic, but not post-operative, pain states[J]. Pain, 2006,123(1-2): 75-82.
[46]DONG X W, GOREGOAKER S, ENGLER H, et al. Small in-terfering RNA-mediated selective knockdown of Na(V)1.8tetrodoloxin-resislant sodium channel reverses mechanical al-lodynia in neuropathic rats[J].Neuroscience, 2007, 146(2):812-821.
[47]JARVIS M F, HONORE P, SHIEH C C, et al. A-803467, apotent and selective Navl.8 sodium channel blocker, attenu-ates neuropathic and inflammatory pain in the rat[J]. Proceed-ings of the National Academy of Sciences of the United Statesof America, 2007, 104(20):8520-8525.
[48]BULAJ G, ZHANG M M, GREEN B R, et al. Synthetic muO-conotoxin MrVIB blocks TTX-resistant sodium channel Navl.8and has a long-lasting analgesic activity[J]. Biochemistry, 2006,45(23):7404-7414.
[49]EKBERG J, JAYAMANNE A, VAUCHAN C W, et al. muO-conotoxin, MrVIB selectively blocks Navl.8 sensory neuronspecific sodium channels and chronic pain behavior withoutmotor deficits[J]. Proceedings of the National Academy of Sci-ences of the United States of America, 2006,103(45):17030-17035.
[50]AKOPIAN A N, SOUSLOVA V, ENGLAND S, et al. Thetetrodotoxin-resistant sodium channel SNS has a specializedfunction in pain pathways[J]. Nature Neuroscience, 1999, 2(6):541-548.
[51]LAIRD J M, SOUSLOVA V, WOOD J N, et al. Deficits invisceral pain and referred hyperalgesia in Navl.8 (SNS/PN3)-null mice[J]. The Journal of Neuroscience: the Official Journalof the Sociely for Neuroscience, 2002, 22(19):8352-8356.
[52]HILLSLEY K, LIN J H, STANISZ A, et al. Dissecting the roleof sodium currents in visceral sensory neurons in a model ofchronic hyperexcitability using Navl.8 and Navl.9 null mice[J].The Journal of Physiology, 2006,576(1):257-267.
[53]DIB-HAJJ S D, TYRRELL L, BLACK J A, et al. NaN, a nov-el voltage-gated Na channel, is expressed preferentially in pe-ripheral sensory neurons and down-regulated after axotomy[J].Proceedings of the National Academy of Sciences of the Unit-ed States of America, 1998, 95(15):8963-8968.
[54]DIB-HAJJ S, BLACK J A, CUMMINS T R, et al. NaN/Navl.9:a sodium channel with unique properties[J]. Trends in Neuro-sciences, 2002, 25(5):253-259.
[55]RUGIERO F, MISTRY M, SAGE D, et al. Selective expressionof a persistent tetrodotoxin-resistant Na+ current and Navl.9subunit in rnyenteric sensory neurons[J]. The Journal of Neuro-science : the Official Journal of the Society for Neuroscience,2003, 23(7):2715-2725.
[56]CUMMINS T R, DIB-HAJJ S D, BLACK J A, et al. A novelpersistent tetrodotoxin-resistant sodium current in SNS-nulland wild-type small primary sensory neurons[J].The Journal of Neu-roscience: the Official Journal of the Society for Neuroscience,1999, 19(24):RC43.
[57]BAKER M D, CHANDRA S Y, DING Y, et al. GTP-inducedtetrodotoxin-resistant Na+ current regulates excitability in mouseand rat small diameter sensory neurones[J]. The Journal of Physi-ology, 2003, 548(2):373-382.
[58]COPEL C, OSORIO N, CREST M, et al. Activation of neu-rokinin 3 receptor inCJ-eases Na(v)1.9 current in enteric neu-rons[J]. The Joumal of Physiology, 2009, 587(7):1461-1479.
[59]DIB-HAJJ S D, TYRRELL L, CUMMINS T R, et al. Twottetrodotoxin -resistant sodium channels in human dorsal rootganglion neurons[J]. FEBS Letters, 1999, 462(1-2):117-120.
[60]PORRECA F, LAI J, BIAN D, et al. A comparison of the po-tential role of the tetrodotoxin-insensitive sodium channels,PN3/SNS and NaN/SNS2, in rat models of chronic pain[J]. Pro-ceedings of the National Academy of Sciences of the UnitedStates of America,1999, 96(14):7640-7644.
[61]AMAYA F. WANG H, COSTIGAN M, et al. The voltage-gat-ed sodium channel Na(v)1.9 is an effector of peripheral in-flammatory pain hypersensitivity[J]. The Journal of Neuroscience:the Official Journal of the Society for Neuroscience, 2006, 26(50):12852-12860.
[62]PFRSSON A K, THUN J, XU X J, et al. Autotomy behaviorcorrelates with the DRG and spinal expression of sodiumchannels in inbred mouse strains[J]. Brain Research, 2009, 1285:1-13.
[63]TATE S, BENN S, HICK C, et al. Two sodium channels con-tribute to the TTX-R sodium current in primary sensory neu-rons[J]. Nature Neuroscience, 1998, 1(8):653-655.
[64]PRIEST B T, MURPHY B A, LINDIA J A, et al. Contributionof the tetrodotoxin -resistant voltage-gated sodium channelNavl.9 to sensory transmission and nociceptive behavior[J].Proceedings of the National Academy of Sciences of the Unit-ed States of America, 2005, 102(26):9382-9387.
[65]YU Y Q, ZHAO F, GUAN S M, et al. Antisense-mediatedknockdown of Na(V)1.8, but not Na(V)1.9, generates inhibitoryeffects on complete Freund's adjuvant-induced inflammatorypain in rat[J]. PloS One, 2011, 6(5):e19865.
[66]VAN GENDEREN P J, MICHIELS J J, DRENTH J P. Heredi-tary erythermalgia and acquired erythromelalgia[J]. American Jo-urnal of Medical Genetics, 1993, 45(4):530-532.
[67]DAVIS M D, O'FALLON W M, ROGERS R S, et al. Naturalhistory of erythromelalgia: presentation and outcome in 168patients[J]. Archives of Dermatology, 2000, 136(3):330-336.
[68]YANG Y, WANG Y, LI S, et al. Mutations in SCN9A, encod-ing a sodium channel alpha subunit, in patients with primaryerythermalgia[J]. Journal of Medical Genetics, 2004, 41(3):171-174.
[69]DIB-HAJJ S D, RUSH A M, CUMMINS T R, et al. Gain-of-function mutation in Navl.7 in familial erythromelalgia in-duces bursting of sensory neurons[J]. Brain: a Journal of Neu-rology, 2005, 128(8):1847-1854.
[70]DRENTH J p, TE MORSCHE R H, GUILLET G, et al.SCN9A mutations define primary erythermalgia as a neuro-pathic disorder of voltage gated sodium channels[J]. The Jour-nal of Investigative Dermatology, 2005, 124(6):1333-1338.
[71]WU M T, HUANG P Y, YEN C T, et al. A novel SCN9A mu-tation responsible for primary erythromelalgia and is resistantto the treatment of sodium channel blockers[J]. PLoS One, 2013,8(1):e55212.
[72]CHOI J S, ZHANG L, DIB-HAJJ S D, et al. Mexiletine-re-sponsive erythromelalgia due to a new Na (v)1.7 mutationshowing use-dependent current fall-off[J]. Experimental Neu-rology, 2009, 216(2):383-389.
[73]FISCHER T Z, GILMORE E S, ESTACION M, et al. A novelNavl.7 mutation producing carbamazepine -responsive ery-thromelalgia[J]. Annals of Neurology, 2009, 65(6):733-741.
[74]DRENTH J P, WAXMAN S G. Mutations in sodium-channelgene SCN9A cause a spectrum of human genetic pain disor-ders[J]. The Journal of Clinical Investigation, 2007, 117(12):3603-3609.
[75]DIB-HAJJ S D, CUMMINS T R, BLACK J A, et al. Fromgenes to pain: Na v l.7 and human pain disorders[J]. Trends inNeurosciences, 2007, 30(11):555-563.
[76]SHEETS P L, JACKSON J 0, WAXMAN S G. et al. A Navl.7channel mutation associated with hereditary erylhromelalgiacontributes to neuronal hyperexcitability and displays reducedlidocaine sensitivity[J]. The Journal of Physiology, 2007, 581(3):1019-1031.
[77]FERTLEMAN C R, FERRIE C D, AICARDI J, et al. Paroxys-mal extreme pain disorder (previously familial rectal pain syn-drome)[J]. Neurology, 2007, 69(6):586-595.
[78]FERTLEMAN C R, BAKER M D, PARKER K A, et al.SCN9A mutations in paroxysmal extreme pain disorder: allelicvariants underlie distinct channel defects and phenotypes[J].Neuron, 2006, 52(5):767-774.
[79]DIB-HAJJ S D, ESTACION M, JARECKI B W, et al. Parox-ysmal extreme pain disorder M1627K mutation in humanNavl.7 renders DRG neurons hyperexcitable[J]. Molecular Pain,2008, 4:37.
[80]JARECKI B W, SHEETS P L, JACKSON J O, et al. Paroxys-mal extreme pain disorder mutations within the D3/S4-S5linker of Navl.7 cause moderate destabilization of fast inacti-vation[J]. The Journal of Physiology, 2008, 586(17):4137-4153.
[81]THEILE J W, JARECKI B W, PIEKARZ A D, et al. Navl.7mutations associateD with paroxysmal extreme pain disorder,but not erythromelalgia, enhance Navbeta4 peptide-meDiatedresurgent sodium currents[J]. The Journal of Physiology, 2011, 589(3):597-608.
[82] ESTACION M, DIB-HAJJ S D, BENKE P J, et al. Navl.7gain-of-function mutations as a continuum: A1632E displaysphysiological changes associated with erythrome,lalgia andparoxysmal extreme pain disorder mutations and producessymptoms of both disorders[J]. The Journal of Neuroscience: theOfficial Journal of the Society for Neuroscience, 2008, 28(43):11079-11088.
[83]COX J J, REIMANN F, NICHOLAS A K, et al. An SCN9Achannelopathy causes congenital inability Lo experience pain[J].Nature, 2006, 444(7121):894-898.
[84]AHMAD S, DAHLLUND L, ERIKSSON A B, et al. A stopcodon mutation in SCN9A causes lack of pain sensation[J]. Hu-man Molecular Genetics, 2007, 16(17):2114-2121.
[85]GOLDBERG Y P, MACFARLANE J, MACDONALD M L, etal. Loss-of-function mutations in the Navl.7 gene, underliecongenital indifference to pain in multiple human populations[J].Clinical Genetics, 2007, 71(4):311-319.
[86]DABBY R, SADEH M, GILAD R, et, al. Chronic non-parox-ysmal neuropathic pain-Novel phenotype of mutation in thesodium channel SCN9A gene[J]. Journal of the Neurological Sci-ences, 2011,301(1-2):90-92.
[87]FABER C G, HOEIJMAKERS J G, AHN H S, et al. Gain offunction Nanul.7 mutations in idiopathic small fiber neuropa-thy[J]. Annals of Neurology, 2012, 71(1):26-39.
[88]MULROY M F. Systemic toxicity and cardiotoxicity from localanesthetics: incidence and preventive measures[J]. Regional An-esthesia and Pain Medicine, 2002, 27(6):556-561.
[89]WALIA K S, KHAN E A, KO D H, et al. Side effects ofantiepileptics--a review[J]. Pain Practice : the Official Journalof World Institute of Pain, 2004, 4(3):194-203.
