多重PCR快速检测恶性疟和间日疟的研究
2014-04-09王晓春
李 欢,孙 菲,文 岚,王晓春
疟疾是最严重的全球性公共健康问题之一,世界上超过一半的人口生活在疟疾流行区。近年来我国输入性疟疾呈逐年上升趋势,2011年[1]全国27个省(市、区)报告病例数4 479例,输入病例占66.4%,死亡病例33例。湖南省报告148例,主要为恶性疟原虫(Plasmodiumfalciparum,P.f)和间日疟原虫(Plasmodiumvivax,P.v)感染。目前我国仍采用显微镜检法作为诊断疟疾的“金标准”,显微镜检法受镜检人员的主观判断及责任心影响,极容易漏诊,延误治疗。为提升对疟疾病例的实验室复核和确诊能力,近年来以PCR为基础的分子生物学诊断技术发展迅速。污染风险低、灵敏度及特异性高的Real-time PCR[2]需要昂贵的仪器及试剂,难以在基层疾控部门推广。本研究通过对多重PCR与传统显微镜检法的比较,旨在建立一种简便快速、适于在基层疾控部门推广的疟疾确诊方法。
1 材料与方法
1.1样本与试剂 本研究采用的样本包括DNA样本和全血标本。恶性疟原虫(3D7)、间日疟原虫(Sal-1)、三日疟原虫(Plasmodium malariae,P.m)(Uganda I)及卵形疟原虫(Plasmodium ovale,P.o)(Nigeria I)DNA由长沙市疾控中心惠赠。从长沙市芙蓉区疾控中心、长沙市疾控中心、中南大学湘雅医院、中南大学湘雅二医院、中南大学湘雅三医院等收集疑似疟疾临床全血标本共86份,血片由湖南国际旅行卫生保健中心同一专家进行显微镜检诊断。DNA提取试剂盒购自QIAGEN公司,PCR试剂盒购自北京康为世纪生物科技有限公司,pGM-T克隆试剂盒与质粒抽提试剂盒购自北京天根生化科技有限公司。
1.2方法
1.2.1引物设计 从GenBank检索P.f和P.v的18S rRNA序列,结合参考文献[3]用Primer 5.0设计引物,并进行BLAST序列比对验证引物特异性。引物由上海生工生物有限公司进行合成及ULTRAPAGE纯化。引物序列见表1。

表1 多重PCR引物序列
1.2.2DNA的提取 吸取100 μL EDTA抗凝全血于1.5 mL eppendorf管中,用QIAGEN的QIAamp DNA Micro Kit试剂盒按照说明书提取DNA,经过裂解、上柱吸附、脱盐等步骤,最后用100 μL elution buffer洗脱DNA。用琼脂糖凝胶电泳法及紫外分光光度法检测所提DNA的纯度及浓度。-20 ℃保存备用。
1.2.3单重PCR 将引物稀释至10 μmoL/L,对引物浓度进行优化,优化后的反应体系为:2×Taq Master Mix(含染料)25 μL,引物PRvf 0.5 μL,引物PRv或PRf 0.5 μL,DNA 5 μL,用RNase-Free ddH2O补足50 μL。对退火温度进行优化,优化后的反应条件为:95 ℃预变性5 min,95 ℃变性30 s,58 ℃退火30 s,72 ℃延伸30 s,循环35次,最后72 ℃延伸5 min,4 ℃结束PCR扩增。取PCR产物6 μL,用25 g /L 琼脂糖凝胶进行电泳,凝胶成像系统中观察扩增结果。回收、纯化PCR产物,送至上海生工生物技术服务有限公司进行测序。
1.2.4多重PCR 将P.f DNA与P.v DNA按照1∶1的比例混合,标为Pvf。对引物浓度进行优化,优化后的反应体系为:2×Taq Master Mix(含染料)25 μL,引物PRvf 1.0 μL,引物PRv 0.5 μL,引物PRf 0.5 μL,DNA 5 μL,用RNase-Free ddH2O补足50 μL。反应条件同单重PCR。
1.2.5最低检测限检测 将P.f、P.v的扩增片段分别插入pGM -T 载体,按试剂盒说明书进行连接、转化和筛选,进行菌落PCR初步验证插入片段是否正确,提取阳性质粒DNA,用紫外分光光度计测定核酸浓度,并计算每微升的拷贝数。以质粒DNA作为标准品,进行10倍梯度稀释。分别取浓度为101~ 107copies /μL 的标准品进行PCR,25 g /L 琼脂糖凝胶电泳进行产物的检测。
1.2.6临床标本的检测 对86例临床全血标本进行多重PCR检测,反应体系与反应条件与上文相同。多重PCR检测结果与湖南国际旅行卫生保健中心的显微镜检结果进行比较。
1.2.7统计学方法 应用SPSS 17.0进行统计数据分析,采用配对四格表资料χ2检验。P<0.05为两种方法有差别。以镜检法为金标准,计算多重PCR检测临床标本的灵敏度等指标。
2 结 果
2.1DNA纯度及浓度 紫外分光光度法测得所有DNA样本A260/A280均在1.8~2.0之间,表明DNA纯度较高。
2.2PCR检测结果 单重PCR可扩增出431 bp(P.f)和341 bp(P.v)目的条带。多重PCR能扩增出模拟混合感染标本Pvf,P.m及P.o无条带,特异性好。见图1。

图1单重与多重PCR产物条带
Fig.1ProductbandsofsinglePCRandmultiplexPCR
1: Single PCRP.f(431 bp); 2: Single PCRP.v(341bp); 3: Multiplex PCRP.f; 4: Multiplex PCRP.v; 5: Multiplex PCR pvf; 6:P.m;
7:P.o; 8: NTC; M: Marker
2.3最低检测限检测 对P.v质粒标准品进行PCR检测的凝胶电泳图显示,体系中标准品浓度为103copies/μL时仍有明显条带,该反应体系中标准品终浓度为102copies/反应。见图2。

图2间日疟原虫梯度稀释标准品的PCR产物条带(341bp)
Fig.2PCRproductbandsofP.vgradientdilutedstandardplasmids
M: Marker; 1: 107copies/μL; 2: 106copies/μL; 3: 105copies/μL; 4: 104copies/μL; 5: 103copies/μL; 6: 102copies/μL; 7: 101copies/μL; 8: NTC.
2.4临床标本检测 对86例临床标本进行检测,两种方法检测结果的比较见表2。镜检法检出40例P.f(见表3)、22例P.v(见表4),多重PCR检出45例P.f、20例P.v。

表2 两种方法检测结果比较
Note: χ2=0.364,P=0.549>0.05,两种方法检测结果无差别。

表3 两种方法检出P.f的比较
Note:P=0.180,P>0.05,两种方法检出P.f的结果无差别
3 讨 论
疟疾是一种极具破坏性的寄生虫病,且近年来P.f对多种抗疟药产生抗药性,宿主蚊亦对杀虫剂产生抗药性。及时、准确的诊断对实现疟疾的控制与消除计划具有重要意义。疟疾的确诊有赖于实验室病原学诊断,传统的镜检法结果受诸多因素影响,容易误诊或漏诊,作为“金标准”已越来越难于评价诊断效率[4]。本研究所采用的多重PCR法可快速检测出是否存在疟原虫感染,并可单管多检,通过一次检测即可鉴别P.f与P.v,两种疟原虫目的片段长度相差90 bp,通过琼脂糖凝胶电泳后容易分辨。操作简便,快速经济,不需要昂贵的仪器与试剂。相对于real-time PCR[5]更适合在基层疾控部门应用。与Singh B等[6]所述的巢式PCR相比较操作更简单、更快速。我国疟疾患者P.f与P.v感染较常见,而P.f对氯喹等耐药,若缺乏及时有效的治疗很容易转化为凶险型发作,危及生命。因此对P.f与P.v进行鉴别对医生正确使用抗疟药,疟疾患者得到有效治疗有着重要的意义。
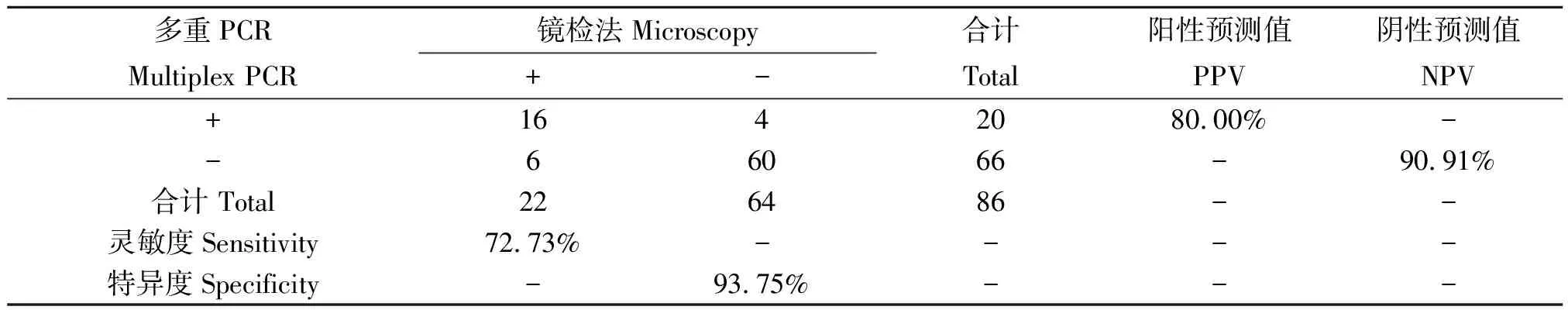
表4 两种方法检出P.v的比较
Note:P=0.754,P>0.05,两种方法检出P.v的结果无差别。
在本研究中,对多重PCR反应的引物浓度和退火温度进行优化,在优化了反应条件下,对4种感染人的疟原虫DNA进行检测,能检出P.f和P.v,不常见的P.o和P.m无特异性条带。对梯度稀释的质粒标准品进行检测,结果显示最低检测限为102copies/反应,与以往报道[7-8]所述多重PCR的最低检测限相同。
对临床标本检测结果显示多重PCR敏感度高,阳性预测值与阴性预测值均大于80%。该方法特异度仅为70.83%,主要是由于多重PCR可检出102copies/反应的疟原虫,但原虫密度低时(<50/μL)镜检法容易漏诊[9]。本研究不足之处在于没有条件对结果不一致的标本用镜检法进行复检。郭传坤[10]等对PCR法与镜检法结果不一致的血片进行复检证实多数属于镜检法的误判。因此,原虫密度低时若只进行显微镜检易漏诊,延误治疗。本研究所采用的多重PCR同时检测P.f和P.v的方法,无需昂贵的仪器与试剂,操作简便、相对快速经济,对这2种疟原虫感染具有确诊价值,为基层疾病控部门进行疟疾的流行病学调查提供手段,有助于及早发现疟疾患者,并准确分型,为临床用药及疗效观察提供参考。
参考文献:
[1]Xia ZG, Yang MN, Zhou SS. Malaria situation in the People’s Republic of China in 2011[J]. Chin J Parasitol Parasit Dis, 2012, 30(06): 419-422. (in Chinese)
夏志贵, 杨曼尼, 周水森. 2011年全国疟疾疫情分析[J]. 中国寄生虫学与寄生虫病杂志, 2012, 30(06):419-422.
[2]Rougemont M, Van Saanen M, Sahli R, et al. Detection of fourPlasmodiumspecies in blood from humans by 18S rRNA gene subunit-based and species-specific real-time PCR assays[J]. J Clin Microbiol, 2004, 42(12): 5636-5643.
[3]Lin M, Gao ST. A field study on detection ofPlasmodiumfalciparumandPlasmodiumvivaxin mixed infection areas by PCR-based method[J]. J Pathog Biol, 2004, 17(03): 39-40. (in Chinese)
林敏, 高世同. PCR检测疟疾混合感染的现场应用研究[J]. 中国寄生虫病防治杂志, 2004,17(03):39-40.
[4]Barber BE, William T, Grigg MJ, et al. Limitations of microscopy to differentiatePlasmodiumspecies in a region co-endemic forPlasmodiumfalciparum,PlasmodiumvivaxandPlasmodiumknowlesi[J]. Malar J, 2013, 12: 8. DOI: 10.1186/1475-2875-12-8
[5]Kaisar MM, Supali T, Wiria AE, et al. Epidemiology ofPlasmodiuminfections in Flores Island, Indonesia using real-time PCR[J]. Malar J, 2013, 12(1): 169.
[6]Singh B, Bobogare A, Cox-Singh J, et al. A genus- and species-specific nested polymerase chain reaction malaria detection assay for epidemiologic studies[J]. Am J Trop Med Hyg, 1999, 60(4): 687-692.
[7]Mixson-Hayden T, Lucchi NW, Udhayakumar V. Evaluation of three PCR-based diagnostic assays for detecting mixedPlasmodiuminfection[J]. BMC Res Notes, 2010, 3: 88. DOI: 10.1186/1756-0500-3-88
[8]Padley D, Moody AH, Chiodini PL, et al. Use of a rapid, single-round, multiplex PCR to detect malarial parasites and identify the species present[J]. Ann Trop Med Parasitol, 2003, 97(2): 131-137.
[9]Berry A, Fabre R, Benoit-Vical F, et al. Contribution of PCR-based methods to diagnosis and management of imported malaria[J]. Med Trop (Mars), 2005, 65(2): 176-183.
[10]Guo CK, Li XM, Lin Z, et al. Primary evaluation on the application of nested/multiplex PCR in malaria diagnosis and surveillance[J]. Chin J Parasitol Parasit Dis, 2008, 26(04): 277-280. (in Chinese)
郭传坤, 黎学铭, 林珍, 等. 套式/多重PCR方法应用于疟疾诊断与监测的初步评价[J]. 中国寄生虫学与寄生虫病杂志, 2008, 26(04):277-280.
