离子对色谱法同时测定益母草片中盐酸水苏碱、盐酸益母草碱
2014-04-02符滇海刘秋雨卢蓉娜
符滇海, 冯 叶, 刘秋雨, 卢蓉娜
(佛山市顺德区药品检验所,广东 顺德 528300)
益母草片为益母草制成的片剂,功能主治活血调经,用于月经量少。目前其质量标准[1]以盐酸水苏碱为定量指标,用重量法测定其含有量。该方法分析步骤多,反应时间长,试验条件严格,大大影响了该检验速度和准确性。除此之外,也有采用高效液相色谱法[2-5],但由于保留时间过短、色谱柱不通用或峰形不理想,加之盐酸水苏碱具有的特殊的分子结构,不适用于益母草片的定量测定。本方法采用C18柱梯度洗脱同时测定盐酸水苏碱和盐酸益母草碱,使用普通试剂,采用独特的样品预处理方法,克服了现有质量标准的弱点。
1 仪器与试药
Dionex U3000高效色谱仪,二极管阵列检测器;昆山KQ-300DA型超声振荡器(功率300 W,频率40 kHz),盐酸水苏碱化学对照品(中国药品生物制品检定所,批号110712-201010,纯度为99.9%),盐酸益母草碱化学对照品(中国药品生物制品检定所,批号111823-201202,纯度为94.7%),益母草片(广东环球制药有限公司,批号分别为101101、110903、110904)。阴性辅料空白(广东环球制药有限公司提供)乙腈为色谱纯,十二烷基硫酸钠(化学纯)、戊烷磺酸钠、己烷磺酸钠、庚烷磺酸钠、辛烷磺酸钠(以上4种离子对试剂均为色谱专用)、磷酸为分析纯,水为超纯水。
2 方法与结果
2.1 色谱条件 Agilent ZORBAX SB-C18色谱柱(250 mm×4.6 mm, 5 μm),流动相为0.010 mol/L辛烷磺酸钠溶液(用磷酸溶液调至pH至2.0)-乙腈,梯度洗脱(0~10 min,10%乙腈;10~15 min,10%~24%乙腈;15~25 min,24%乙腈;35~40 min,4%~10%乙腈,40~50 min,10%乙腈);检测波长为192 nm(盐酸水苏碱),218 nm(盐酸益母草碱);体积流量为1.0 mL/min;柱温35 ℃;进样量10 μL。
2.2 供试品溶液制备 取10片益母草片,除去包衣,精密称定,研细,精密称定1.972 8 g(约相当于盐酸水苏碱45 mg),置100 mL量瓶中,70%乙醇约70 mL,超声45 min,放冷,加70%乙醇至刻度,摇匀,滤过,精密量取续滤液5 mL,加在酸性氧化铝柱(200~300目,5 g,内径为1.5 cm,湿法装柱)上,用水20 mL洗脱,收集洗脱液于蒸发皿中,置水浴上蒸干,用70%乙醇溶液溶解并转移至5 mL的量瓶中,定容,摇匀,滤过,即得。
2.3 对照品制备 取盐酸水苏碱、盐酸益母草碱对照品适量,精密称定,加70%乙醇溶解,分别制成每1 mL约含0.25 mg、7.0 μg和0.35 mg、9.0 μg的两份混和对照品溶液。用两点校正法计算。
2.4 线性关系 精密称取盐酸水苏碱对照品适量,用70%乙醇溶解得到质量浓度为914.5 μg/mL贮备液,分取制得18.29、36.57、91.43、182.86、274.30、548.58、914.30 μg/mL的对照品溶液,按“2.1”项下的色谱条件进行测定。在上述色谱条件下分别进样10 μL,以质量浓度(x)为自变量,峰面积(y)为因变量,得到回归方程为y=1.720 3x+3.213,r=0.999 96(n=7)。结果盐酸水苏碱在18.29~914.30 μg/mL范围内与峰面积呈良好的线性关系。
精密称取盐酸益母草碱对照品适量,用70%乙醇溶解得到质量浓度为32.26 μg/mL贮备液,分取制得0.645 3、1.613、3.226、6.453、12.91、19.36、32.26 μg/mL的对照品溶液,按“2.1”项下的色谱条件进行测定。在上述色谱条件下分别进样10 μL,以质量浓度为自变量,峰面积为因变量,得到回归方程y=44.86x-4.70,r=0.999 97(n=7)结果盐酸益母草碱在0.645 3~32.26 μg/mL范围内与峰面积呈良好的线性关系。
2.5 精密度试验 取“2.3”项下的对照品溶液,按“2.1”项下的色谱条件进行测定,重复测定6次,按峰面积及出峰时间计算精密度,盐酸水苏碱峰面积的RSD为0.2%,保留时间的RSD为0.04%,盐酸益母草碱峰面积的RSD为0.3%,保留时间的RSD为0.04%,表明仪器精密度良好。
2.6 稳定性试验 取同一批号供试品溶液置于室温,分别于0、2、4、6、8、14 h内按“2.1”项下的色谱条件进行测定。以峰面积计算盐酸水苏碱和盐酸益母草碱成分相对标准偏差RSD分别为0.7%、0.6%,表明供试品溶液在14 h内基本稳定。
2.7 重复性试验 精密称取同一批号(广东环球制药有限公司,批号110904,含盐酸水苏碱15 mg/片)分别按“2.2”项下的方法平行制备6份供试品溶液,按“2.1”项下的色谱条件进行测定,盐酸水苏碱为标示量59.6%,RSD为0.9%。盐酸益母草碱0.288 mg/片,RSD为0.5%。
2.8 回收率试验 精密称取已知含有量的样品(广东环球制药有限公司,批号110904,含盐酸水苏碱15 mg/片)9份,折合为盐酸水苏碱和盐酸益母草碱的量,置25 mL的量瓶中,并精密加入不同体积的同一质量浓度的对照品溶液,按“2.2”项下的方法处理,加70%乙醇稀释至刻度,摇匀,制成不同的供试品溶液。按“2.1”项下的色谱条件进行测定,结果盐酸水苏碱和盐酸益母草碱的平均回收率分别为99.7%(RSD 1.4%)、98.2%(RSD 0.7%)。见表1、2。
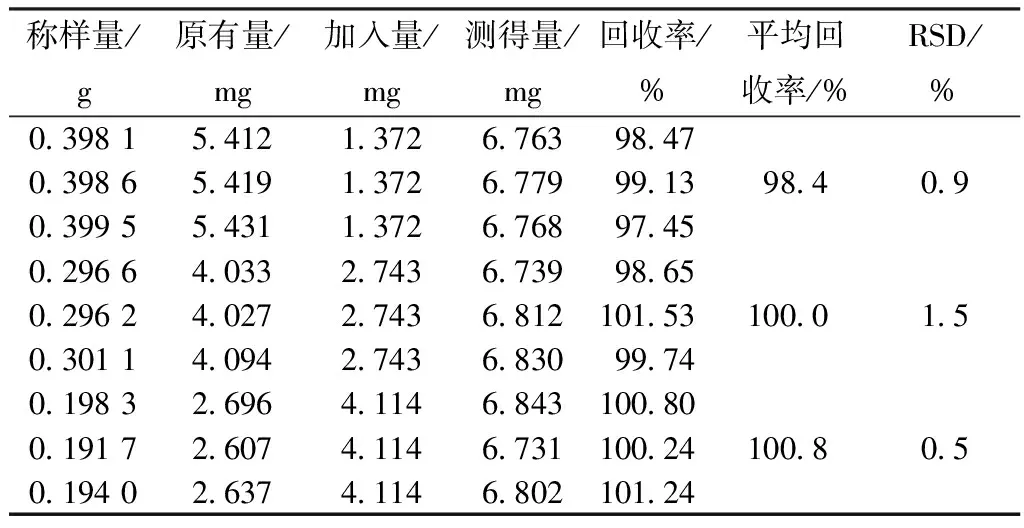
表1 盐酸水苏碱加样回收率试验
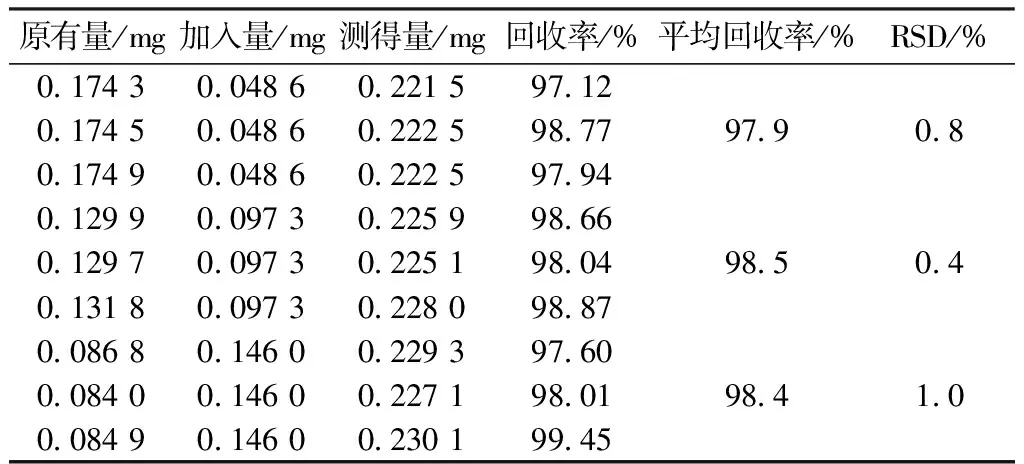
表2 盐酸益母草碱加样回收率试验
2.9 样品测定 取样品(广东环球制药有限公司,批号110904:含盐酸水苏碱15 mg/片),按“2.2”项下的方法制备供试品溶液,按“2.1”项下的色谱条件进行测定,以外标法计算盐酸水苏碱的量。色谱图见图1、图2。
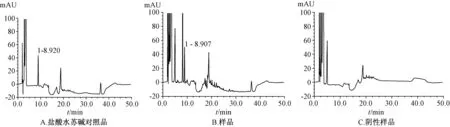
图1 盐酸水苏碱谱图(192 nm)
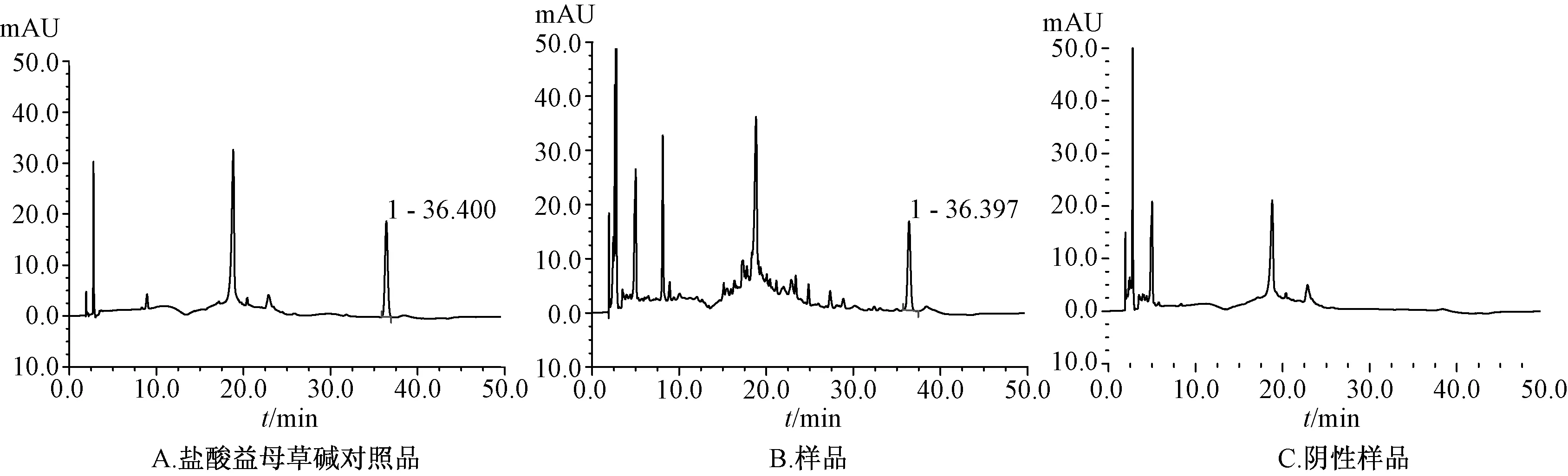
图2 盐酸益母草碱色谱图(218 nm)
用同样的方法对该品种项下的3个批次(101101,110903,110904)样品进行了测定。结果盐酸水苏碱分别为标示量65.1%(n=3,RSD为0.9%)、58.2%(n=3,RSD为0.7%)、59.6%(n=6,RSD为0.9%);盐酸益母草碱分别为0.278 mg/片(101101)(n=3,RSD为0.4%)、0.283 mg/片(110903)(n=3,RSD为0.5%)、0.288 mg/片(110904)(n=6,RSD为0.5%)。而用国家标准方法[1]测定得盐酸水苏碱的百分标示量分别为100.8%(101101)、98.3%(110903)和98.2%(110904)(该数据由广东环球制药有限公司提供)(标准规定,每片含盐酸水苏碱(C7H13NO2·HCl)应为标示量的90.0~110.0%)。由此可见,本方法与国家药品标准方法相比,测定结果有较大的出入。
3 讨论
3.1 流动相离子对试剂的选择 将十二烷基硫酸钠、戊烷磺酸钠、己烷磺酸钠、庚烷磺酸钠、辛烷磺酸钠配成相同摩尔浓度的离子对溶液,在相同p H2的条件下,按相同流动相比例进样,记录色谱图。结果发现:所有离子对试剂都能使样品出现色谱峰戊烷磺酸钠、己烷磺酸钠作为离子对试剂,盐酸水苏碱保留时间过早;十二烷基硫酸钠在此流动相比例下不能完全溶解,且过滤后的流动相保留时间过长,峰形拖尾;辛烷磺酸钠较庚烷磺酸钠保留时间较长,有利于流动性比例的调整。且辛烷磺酸钠作为流动相基线较稳,空白进样在主峰相应的位置上无干扰;当水系流动相中辛烷磺酸钠的浓度为10 mmol/L时,盐酸水苏碱色谱峰的理论塔板数和对称性较理想。且该色谱条件与《中国药典》2010年版[2]收载的盐酸益母草碱方法相近,所以通过梯度洗脱可完成盐酸水苏碱和盐酸益母草碱的同时测定,效果良好。
3.2 流动相的pH值选择 以10 mmol/L辛烷磺酸钠溶液-乙腈(90∶10)为流动相,将水系和有机系混合后,用磷酸调pH,考察了pH从5.0~2.0对盐酸水苏碱色谱图的色谱行为的影响,随着pH的降低,保留时间也随着增加,在pH约为2时,保留时间变得稳定。结果发现,当pH在2时,在此条件下,能很好地满足分析要求。
3.3 样品的制备 我们参考了《中国药典》[2]的提取方法并结合了药品的制备工艺,采用70%乙醇为提取溶剂,和原标准中以0.1 moL/L盐酸提取效果一样,超声提取30 min与回流提取相同时间效果也一致,且浓度不再增加,因此采取提取时间为45 min比较合适。采用氧化铝过柱,以水为洗脱液,盐酸水苏碱、盐酸益母草碱对照品溶液无损失,样品的回收率均很高,且能去除大部分干扰杂质。试验发现,使用酸性氧化铝去除杂质效果最佳。
3.4 方法耐用性考察 对不同厂牌型号的色谱柱进行了考察,其中有Agilent ZORBAX SB-C18(250 mm×4.6 mm,5 μm)、phnomenex Luna 5 μm C18(250 mm×4.6 mm,5 μm)、Syncronis C18(250 mm×4.6 mm,5 μm),均能得到柱效高,对称性较好的色谱图,且峰面积基本一致。表明本方法通用性、耐用性、重复性均良好。
3.5 测定结果讨论 该方法与国家标准方法[1]测定存在较大的出入。究其原因,在国家标准中使用的与盐酸水苏碱起沉淀反应的试剂硫氰酸铬铵溶液,可能与药品中的其他物质也产生的沉淀反应,致使其比用高效液相法测得的值高出许多。同时还考察了按国家标准方法(10片置50 mL的量瓶中)与本实验“2.2”项供试品制备进行了比较,发现用本方法测得的含有量结果基本一致。说明国家标准以盐酸水苏碱作为质量指标存在较大误差。
参考文献:
[1] 国家药典委员会.药典业发字(2000)第231号[S].
[2] 国家药典委员会.中华人民共和国药典:2010年版一部[S].北京:中国医药科技出版社,2010:272.
[3] 戚建中.产妇康颗粒中盐酸水苏碱HPLC分析[J].中成药, 2001,23(1):16-18.
[4] 杨晓云,扆雪涛,张云丽.益母草丸中盐酸水苏碱成分分析方法建立[J].中国现代应用医学杂志, 2008,25(1):69-70.
[5] 严优芍,李卫民.HPLC法测定益母草分散片中盐酸水苏碱的含量[J].广东药学院学报,2006,22(6):604-606.
[6] 任江剑,徐建忠,王志安.HPLC法测定鲜益母草中盐酸水苏碱含量[J].中药新药与临床药理,2006,17(3):216-218.
[7] 高晓波,张小勇,黄春清.HPLC法测定益母草中盐酸水苏碱含量的色谱条件研究[J].中医药信息,2009,26(4):25-27.
[8] 余 琪,章署丹.益母草片中盐酸水苏碱的HPLC测定[J].中国医药工业杂志,2006,37(1):34-35.
[9] 张 韵,黄瑞红,何桂区.HPLC法测定安坤益母草片中盐酸水苏碱的含量[J]. 中成药,2008,24(1):16-17.
[10] 李政海,苏作林,廖展苇,等.HPLC法测定益母草颗粒(无糖型)中盐酸水苏碱的含量[J]. 药品鉴定,2011,8(16):70-72.
[11] 宁康健,冯惠平,何胜利.高效液相色谱法测定前列泰胶囊中盐酸水苏碱的含量[J].中国实验方剂学杂志,2006,12(9):9-11.
[12] 程立方,崔秀君.产舒康颗粒中盐酸水苏碱高效液相色谱法测定[J].中国现代应用药学杂志,2007,24(6):496-498.
[13] 邹洪君,王宇杰,王忠海.高效液相色谱法测定益母草胶囊中盐酸水苏碱含量[J].辽宁中医杂志, 2008,35(7):1064-1065.
[14] 唐 哲,刘 丽,张 玮.RP-HPLC法测定益母草不同部位盐酸水苏碱的含量[J].中国中医药信息杂志, 2008,15(8):51-52.
[15] 林家寿.不同地区益母草的水苏碱含量比较[J].中国医药指南, 2011,9(8):32-33.
[16] 周立伟,张 赞.HPLC法测定益母草片中盐酸水苏碱的含量[J].中国现代中药,2011,13(7):33-34.