原发性低钾型周期性麻痹患者的基因及影像学研究
2014-03-30贾白雪杨旗王涵郭秀海
贾白雪 杨旗 王涵 郭秀海
原发性低钾型周期性麻痹(hypokalemic periodic paralysis, HypoPP )是一种以发作性的迟缓性瘫痪伴血钾水平降低为特征的骨骼肌离子通道病[1]。在高加索人种中,多数HypoPP患者为家族性(familial periodic paralysis, FPP),呈常染色体显性遗传,涉及的基因有CACNA1S基因和SCN4A基因。有近90%的HypoPP患者被发现在上述基因编码骨骼肌离子通道电压感受器S4片段的外显子上存在导致精氨酸替换的点突变[2]。亚洲各国报道的HypoPP患者以散发性(sporadic periodic paralysis, SPP)为主,少数以家系形式存在,多数患者在上述两个基因上未发现突变[3-6]。
近年来,肌肉MRI由于其无创、清晰及可重复等特点而被广泛应用于神经肌肉疾病的诊断和评估。本研究观察了中国HypoPP患者的基因特点,并分析了HypoPP患者发作间期的小腿肌肉MRI表现及病变分布特点。
1 对象和方法
1.1对象收集就诊于首都医科大学宣武医院神经内科门、急诊,确诊为原发性HypoPP的患者共40例,年龄15~61岁,平均(31.0±7.9)岁,包括SPP患者33例,FPP患者7例(其中1例来自于一个四代家系,其余6例均为仅父亲有类似发作史)。选取10例年龄匹配的健康对照者。入组标准:(1)有发作性骨骼肌弛缓性瘫痪而无感觉障碍;(2)至少有一次发作时进行血清钾检测,血钾水平低于3.5 mmol/L;(3)补钾治疗有效;(4)有或无类似发作的家族史。排除标准:(1)甲状腺功能亢进,不能除外甲亢性周期性麻痹者;(2)存在肾小管酸中毒、糖尿病酸中毒、糖皮质激素使用等其他导致低血钾者;(3)有MRI检查禁忌的患者;(4)拒绝基因筛查及MRI检查者。所有受试者均签署知情同意书。
上述一四代家系共有9例患者,其中1例已去世(图1)。首次发病年龄10~30岁,其中6例为20岁左右发病,50岁以后逐渐停止发作;发病诱因有受凉等,部分患者发病无明确诱因;无力以双下肢为主,重者可出现四肢无力,发作频率每个月数次至每年一到两次,部分患者发作时测血钾降低,补钾治疗有效。
1.2方法
1.2.1热点基因筛查:取外周血3 mL,应用高盐沉淀法提取基因组DNA[7]。
1.2.2PCR检测:CACNA1S基因第4、11、21、30外显子和SCN4A 基因第5、12、13、18、24外显子为编码电压感受器的部分。利用9对引物[2],应用PCR技术扩增上述9个外显子,引物由上海生工生物工程有限公司合成。(1)扩增CACNA1S基因各外显子的PCR:反应体系30 μL,含DNA 200 ng,10×PCR Buffer 5 μL,10 mol/L上下游引物各2 μL,dNTPs 200 μmol/L,TaqDNA聚合酶2.5 U(日本TaKaR公司)。(2)扩增SCN4A基因各外显子的PCR:反应体系50 μL ,含DNA 200 ng ,10×PCR Buffer5 μL, 10 mol/L上下游引物各1.5 μL ,dNTPs 200 μmol/L,TaqDNA聚合酶1.0 U(日本TaKaR公司)。 PCR反应条件:采用PTC-100 PCR仪(美国BIO-RAD),预变性95℃ 10 min×1个循环;变性95℃ 30 s,退火60℃(SCN4A基因为58℃)30 s,延伸72℃ 30 s×30个循环;延伸7 min×1个循环。应用2%(质量浓度)琼脂糖电泳检测PCR产物的质量,用密尼博纯化版纯化。

:先证者;□:健康男性;○:健康女性;■:男性患者;●:女性患者;:死亡
图1HypoPP患者一家系遗传图谱
1.2.3基因测序:用美国产ABI 3730XL DNA测序仪对纯化后PCR产物进行正反向测序,明确有无突变。测序由赛诺基因公司完成。对发现阳性突变的家族性HypoPP患者,对其家系中其余患者及部分未发病者进行基因筛查。
1.2.4双下肢MRI检查:对20例患者(FPP患者4例,SPP患者16例,于发作间期进行)和10例健康受试者进行双小腿MRI检查。采用SIMENS 3.0T MR机,分别进行轴位及冠状位脂肪抑制T2WI扫描,参数为T2-TSE-TRA-FS(TR=4540 ms,TE=109 ms,ST=5.0 mm)及T2-TSE-COR-FS(TR=3670 ms,TE=95 ms,ST=5.0 mm)。所有受试者在检查前2 d避免剧烈运动。影像学量化方法:(A):由一名临床医师和一名影像科医师进行测量,采用盲法对每个患者及健康受试者的轴位脂肪抑制T2WI选取3个层面,在各层面上共同选取感兴趣区域(region of interest,ROI),测量信号强度,并以相应区域的背景信号强度为标准进行标准化,将3个层面的平均值作为该患者的信号强度值,以此代表其肌肉水肿程度。(B):由一名影像专业医师按照下述标准对所有影像的每块肌肉进行盲法评分[8]:0分:正常表现——肌肉MRI上无异常信号;1分:轻度异常——散在的斑片状高信号,异常信号小于肌肉体积的30%;2分:中度异常——片状高信号,异常信号占肌肉体积的30%~60%;3分:重度异常——肌肉60%以上的区域显示异常高信号,或肌肉与周围肌肉间分界不清,难以辨认。每块肌肉以每例患者(双侧)的受累次数(0~2次)作为该肌肉是否易受累的评价指标,以每例患者(双侧)的评分(0~6分)作为该肌肉受累程度的评价指标。
1.3统计学处理采用SPSS16.0进行数据分析,符合正态分布的计量资料以均数±标准差表示,采用t检验;非正态分布数据采用中位数和四分位数间距间距表示,采用Kruskal-Wallis检验。以P<0.05表示有统计学差异。
2 结果
2.1基因型及表型基因筛查共发现阳性突变2例,基因突变均位于CACNA1S基因上,分别为1例SPP患者(R1239H)和前述一四代家系的患者(R900S)。余38例HypoPP患者(FPP患者6例,SPP患者32例)均未在上述基因筛查中发现突变。
2.1.1存在R1239H突变SPP患者:患者女,22岁,8岁起病,受凉、进食碳水化合物及情绪紧张时易发作,表现为四肢无力,严重时平躺不能翻身,约1~2个月发作1次。19岁时因近1年发作频繁(每周1次)首次就诊,发作时测血钾降低,补钾治疗有效。否认家族史。目前患者出现双下肢持续性无力、沉重感。患者CACNA1S基因第30外显子上存在c.3716 G>A杂合突变,导致氨基酸R1239H 改变。对其哥哥(健康人)进行基因筛查,相应位点无该突变(图2)。
2.1.2存在R900S突变的一四代家系:对家系中存活的所有患者(8例)及3例未发病亲属(Ⅲ8、Ⅲ11、Ⅳ1)进行了基因筛查。8例患者的CACNA1S基因第21外显子上均发现c.2700 G>C杂合突变,导致氨基酸R900S 改变。3例未发病亲属年龄均小于15岁,其中1例(Ⅲ11,14岁)存在该突变,另外两位家庭成员未见该突变(图3)。
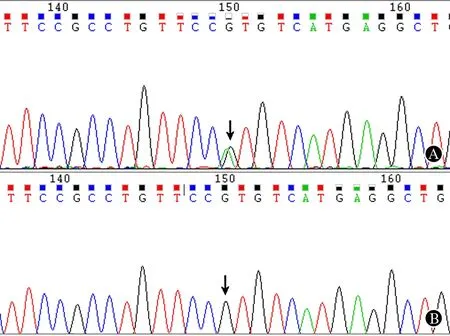
A:患者正向测序结果显示CACNA1S基因CDS位点3716(G→A)杂合突变(↓),导致CGT(Arg)→CAT(His),引起氨基酸改变R1239H;B:患者哥哥(健康人)正向测序结果显示3716 G(↓)无突变,CGT编码精氨酸(Arg)
图2某例SPP患者及其兄的CACNAIS基因第30外显子的测序图谱

A:患者正向测序结果显示CACNA1S基因CDS位点2700(G→C)杂合突变(↓),导致ACC(Arg)→AGC(Ser),引起氨基酸改变R900S;B:正常家庭成员中Ⅲ8正向测序结果正常,为2700G(↓),AGG编码精氨酸(Arg)
图3一家系HypoPP患者及其正常家系成员的CACNA1S基因第21外显子的测序图谱
2.2影像学表现
2.2.1脂肪抑制T2WI表现:20例患者双小腿在脂肪抑制T2WI上均存在不同程度的高信号(标准化信号强度389.7±68.1),10例正常受试者双小腿肌肉在脂肪抑制T2WI上均表现为均匀一致等信号(标准化信号强度275.4±33.2),两者之间存在统计学差异(P<0.01)(图4)。
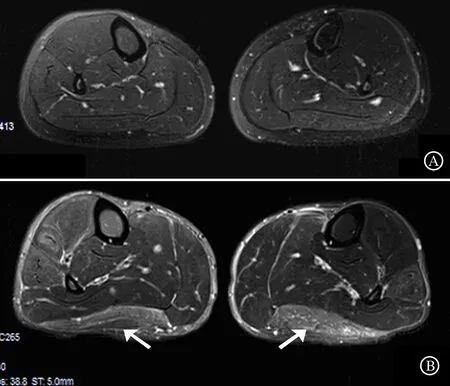
A:健康受试者表现为双小腿肌肉均匀一致等信号;B:患者双侧腓肠肌外侧头呈显著高信号(箭头所示)
图4正常受试者及HypoPP患者小腿MRI(脂肪抑制T2WI)表现比较
2.2.2存在R1239H基因突变患者的MRI表现:R900S突变家系患者未接受MRI检查。存在R1239H突变的1例SPP患者双小腿肌肉受累较重(图5),评分为54分,明显高于另19例患者(评分1~17分,中位数5分,四分位数间距为4分)。
通过对存在R1239H突变的1例SPP患者进行生活指导,嘱其避免诱因,并进行口服补钾治疗,3年后此例患者发作频率明显下降,由每周1次下降至每2~3个月1次,复查下肢MRI可见脂肪抑制T2WI高信号较前明显减轻、局限(图5)。肌肉评分由54分下降至38分。

图5存在R1239H突变SPP患者改变生活方式后3年(B)复查MRI可见左下肢脂肪抑制T2WI高信号较改变生活方式前(A)明显减轻、局限
2.2.3患者肌肉受损分布:患者肌肉受累符合一定的分布规律,最易受累的肌肉分别为腓肠肌外侧头、比目鱼肌、腓肠肌内侧头及胫骨前肌,而腓骨长短肌、胫骨后肌、趾、踇长伸肌及趾踇长屈肌则较少受累(表1)。患者各肌肉受累次数及评分间比较均具有统计学差异(P<0.01)。

表 1 患者小腿肌肉受累次数及受累评分表
3 讨论
该研究结果显示,同其他亚洲国家相似,中国HypoPP患者以SPP为主,呈典型常染色体显性遗传的多代家系仅有1例;共发现两例患者呈阳性突变,分别为位于CACNA1S基因的R1239H(SPP患者)及R900S(四代家系)突变,余患者均未发现突变,表明多数中国HypoPP患者并非由目前已知区域的突变导致,推测可能是位于上述基因其他位点的突变或未知基因的突变引起。
HypoPP的发病机制尚不肯定,目前认为由上述两基因突变导致的精氨酸替换而产生的“门控漏电流”为致病的关键[9]。该电流的持续存在导致骨骼肌细胞膜静息电位稳态改变,在导致血钾降低的诱因的存在下,出现无力的发作。此外,该电流的长期作用使肌细胞内钠离子持续超载,产生细胞毒性,即使在发作间期,患者细胞也可能存在水肿和变性[10-11]。
近年来,人们开始应用MRI,通过对肌肉水分、钠及脂肪的含量及分布的显示来研究HypoPP的发病机制[10]。23Na-MRI是一项能显示骨骼肌内钠离子分布的磁共振序列,H1-MRI能够清晰地显示软组织结构,较稳定地反映肌肉的脂肪变性及水肿病变。Jurkat-Rott 等研究表明,H1-MRI显示的肌肉水肿程度与23Na-MRI显示的钠超载呈线性相关(r2=0.63),认为H1-MRI可作为反映肌细胞内钠超载的手段[10]。
该研究对20例HypoPP患者进行了双下肢MRI检查发现,患者发作间期小腿肌肉MRI的脂肪抑制T2WI信号强度与健康受试者比较差异有统计学意义,表明患者小腿肌肉即使在发作间期存在水肿,这与HypoPP的门控孔道电流导致钠超载理论一致,证实了尽管HypoPP为发作性病程,发作间期患者的表现可完全正常,但其膜内外离子的异常分布持续存在。发作间期患者处于亚临床状态,当有诱因存在时可导致患者无力发作。因此,尽管多数中国HypoPP患者不存在已知位点的突变,但其发病机制与漏电流及钠超载机制存在密切联系。
存在R1239H突变的患者小腿肌肉受累程度严重,即使治疗好转后,其受累评分仍明显高于其他未发现突变患者。由此推测,存在相关位点的经典突变的HypoPP患者其肌膜内外离子的异常分布及钠超载较其余患者严重,这与其临床表现的严重程度一致。
目前对各种神经肌肉疾病的肌肉MRI表现研究较多[12-13],但对HypoPP患者的研究较少,对其下肢肌肉受损的分布特点尚未见报道。该研究发现,HypoPP患者易受累的肌肉为小腿三头肌(比目鱼肌、腓肠肌外侧头及腓肠肌内侧头),均为负重的大块肌肉,而余小肌肉群则较少受累,推测其原因可能是由于负重的肌肉较小肌肉电活动更频繁,肌膜上漏电流更活跃,进而导致钠超载及水肿程度更重,也可进一步继发肌纤维损伤。
此外,对R1239H突变的散发性患者的影像学随访表明,HypoPP患者下肢肌肉MRI改变具有一定可逆性,提示下肢肌肉MRI可能作为治疗效果的监测手段,但其确切结论还需进一步证实。
[1]刘海玲, 童晓欣. 低钾型周期性麻痹的研究进展[J]. 卒中与神经疾病, 2010,17:306-309.
[2]Matthews E, Labrum R, Sweeney M G, et al. Voltage sensor charge loss accounts for most cases of hypokalemic periodic paralysis[J]. Neurology, 2009,72:1544-1547.
[3]Sung CC, Cheng CJ, Lo YF, et al. Genotype and phenotype analysis of patients with sporadic periodic paralysis[J]. Am J Med Sci, 2012,343:281-285.
[4]Wang W, Jiang L, Ye L, et al. Mutation screening in Chinese hypokalemic periodic paralysis patients[J]. Mol Genet Metab, 2006,87:359-363.
[5]Phakdeekitcharoen B, Ruangraksa C, Radinahamed P. Hypokalaemia and paralysis in the Thai population[J]. Nephrol Dial Transplant, 2004,19:2013-2018.
[6]柯青, 吴卫平, 徐全刚, 等. 家族性低钾型周期性麻痹的基因突变与临床特征[J]. 中华神经科杂志, 2006,39:323-327.
[7]吴清敏, 刘巧红, 滕云, 等. 外周血DNA提取方法的比较[J]. 中华检验医学杂志, 2004,26:50-51.
[8]Mercuri E, Talim B, Moghadaszadeh B, et al. Clinical and imaging findings in six cases of congenital muscular dystrophy with rigid spine syndrome linked to chromosome 1p (RSMD1)[J]. Neuromuscul Disord, 2002,12:631-638.
[9]Cannon SC. Voltage-sensor mutations in channelopathies of skeletal muscle[J]. J Physiol, 2010,588(Pt 11):1887-1895.
[10]Jurkat-Rott K, Weber MA, Fauler M, et al. K+-dependent paradoxical membrane depolarization and Na+overload, major and reversible contributors to weakness by ion channel leaks[J]. Proc Natl Acad Sci USA, 2009,106:4036-4041.
[11]Jurkat-Rott K, Groome J, Lehmann-Horn F. Pathophysiological role of omega pore current in channelopathies[J]. Front Pharmacol, 2012,3:112.
[12]Carlier PG, Mercuri E, Straub V. Applications of MRI in muscle diseases[J]. Neuromuscul Disord, 2012,22(Suppl 2):S41.
[13]Tasca G, Iannaccone E, Monforte M, et al. Muscle MRI in Becker muscular dystrophy[J]. Neuromuscul Disord, 2012,22 (Suppl 2):S100-S106.
