食欲素与血糖和能量代谢的调节
2014-03-27综述陆颖理审校
翁 盼(综述),陆颖理(审校)
(1.上海交通大学医学院,上海 200011; 2.上海交通大学医学院附属第九人民医院内分泌代谢科,上海 200011)
食欲素是在1998年首先被发现的,包括食欲素A(含33个氨基酸残基)和食欲素B(含28个氨基酸残基),是由同一前体蛋白裂解产生的一对兴奋性神经肽激素,食欲素由下丘脑中特定的神经元产生,主要位于外侧下丘脑区、穹窿周围区,以及下丘脑后部[1]。在过去的十年里,许多实验已经证实,食欲素在调控一系列生理活动中扮演重要角色,包括调节进食、睡眠、觅食和能量血糖动态平衡等。该文就食欲素与血糖、能量代谢调节和肥胖的研究进展予以综述。
1 食欲素与血糖调节和胰岛素抵抗
1.1血糖的来源与调节因素 血糖的主要来源有食物和肝脏的内源性葡萄糖生成,前者提升血糖的程度主要由食物成分决定,后者主要受胰岛素和胰高血糖素的调节,但也有一部分直接受自主神经支配,经下丘脑控制。胰岛素和副交感神经增加肝脏葡萄糖摄取和糖原合成,而胰高血糖素和交感神经促进糖原分解、糖异生和葡萄糖释放。血糖水平也受肌肉和脂肪组织摄取葡萄糖的影响。
1.2食欲素及受体的分布与作用特征 食欲素是下丘脑血糖调节的重要因子,食欲素纤维分布于整个脑干,包括迷走神经背核和孤束核。食欲素神经元受谷氨酸、促肾上腺皮质素释放因子,神经降压素,血管加压素、催产素和促甲状腺素释放激素的兴奋,而γ-氨基丁酸(GABA)、5-羟色胺、去甲肾上腺素、多巴胺和神经肽Y对食欲素神经元有抑制作用[1]。外周的代谢信号也影响食欲素神经元的活性,葡萄糖和瘦素抑制食欲素神经元的活性,而生长激素促分泌素受体内源性配体则促进其活性。
食欲素主要通过两个G蛋白偶联受体,即食欲素受体1(orexin receptor 1,OXR1)和食欲素受体2(orexin receptor 2,OXR2)发挥作用,OXR1与食欲素A结合的亲和力比食欲素B高100倍,而OXR2与两者结合的亲和力基本相同。下丘脑腹内侧核是OXR1 mRNA最丰富的区域,而OXR2 mRNA高表达于下丘脑结节乳头核、室旁核、弓状核以及外侧下丘脑区。
1.3食欲素与血糖稳态的中枢性调节机制
1.3.1食欲素与血糖的双重调节 下丘脑食欲素系统参与调节血糖稳态:在这个系统中,食欲素神经元精确感知外周营养状态,调节自主神经平衡,控制外周组织产生和利用葡萄糖。下丘脑食欲素神经元能刺激交感神经节前纤维和副交感神经元,通过改变自主神经平衡来对血糖稳态进行双重调节(图1)[2]。
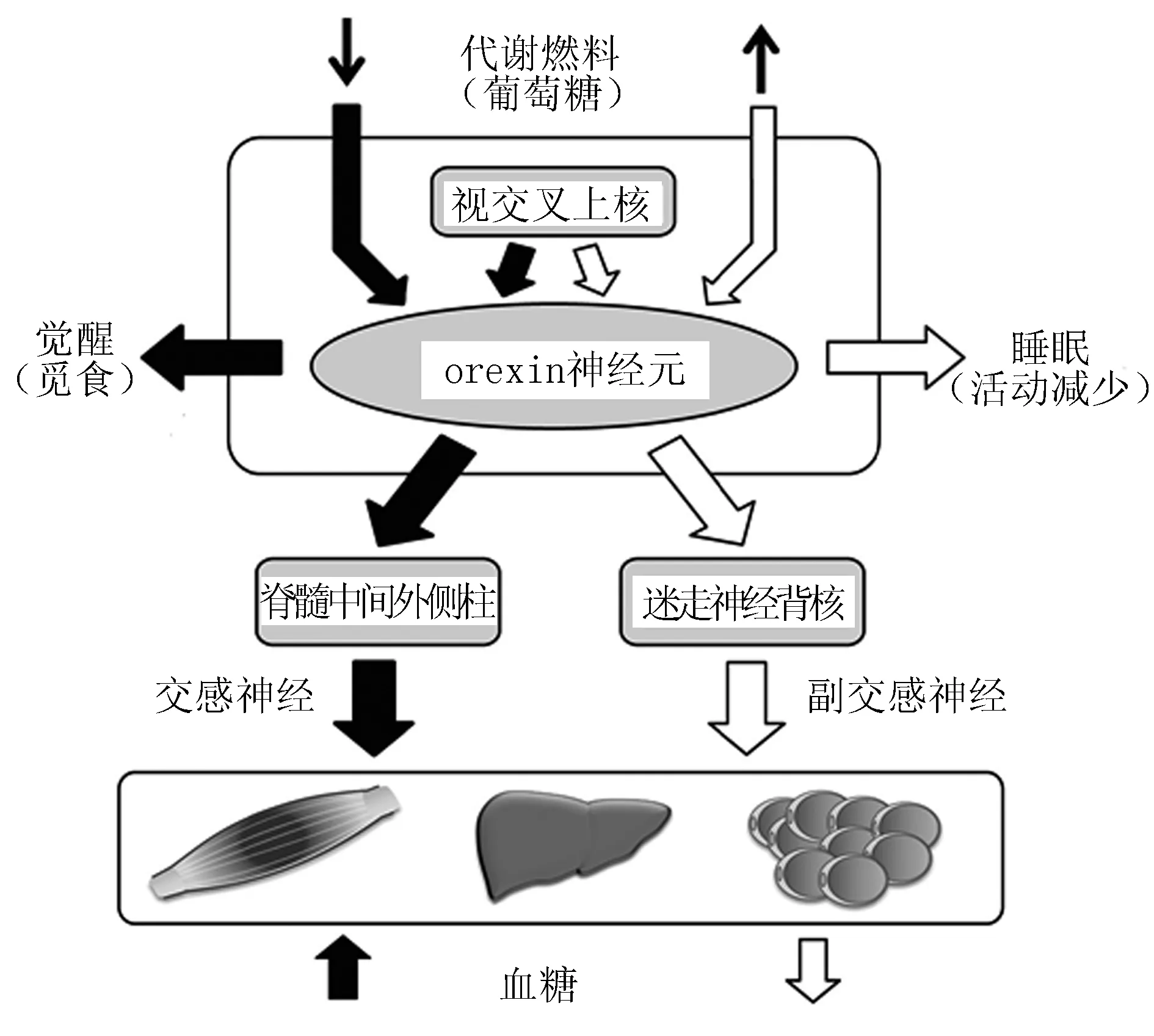
图1 食欲素与血糖稳态双重调节
研究发现,食欲素A可直接激活腹内侧下丘脑神经元,促进交感神经系统激活,然后经由骨骼肌细胞内的β2肾上腺素受体信号介导,增加骨骼肌摄取葡萄糖,促进胰岛素诱导的葡萄糖摄取和肌糖原合成[3-4]。
Ramadori等[5]通过应用一种在下丘脑腹内侧核神经元SF1缺乏食欲素代谢传感器烟酰胺腺嘌呤二核苷酸(NAD+)依赖的脱乙酰酶1(sirtuin type 1,SIRT1)的小鼠(Sf1-Cre;Sirt1loxP/loxP mice)来研究经腹内侧核调控血糖平衡的机制。当给予高热量饮食后,与正常小鼠相比,Sf1-Cre;Sirt1loxP/loxP小鼠体质量增加更显著,能量消耗减少,瘦素抵抗增加。另外,高热量饮食喂养的基因突变小鼠(Sf1-Cre;Sirt1loxP/loxP mice)在出现严重肥胖之前就已经呈现出高胰岛素血症、糖耐量受损和胰岛素抵抗。对于高热量饮食喂养的对照组小鼠,持续侧脑室注射食欲素 A(0.5 nmol/d)能改善高血糖和高胰岛素血症,但在上述基因突变的小鼠中未观察到这种改善现象。这些实验结果提示,下丘脑腹内侧核是食欲素调控肌肉葡萄糖利用的主要部位。
另一方面,食欲素对肝脏葡萄糖生成的调节可能一部分是通过调控人体生物钟完成的。生物钟位于下丘脑视交叉上核,而下丘脑室旁核控制着交感和副交感神经的活动,后者是生物钟信号输出的一个重要靶点,视交叉上核直接控制着血浆葡萄糖浓度的日常节律改变,且不受进食的影响。空腹大鼠室旁核内应用GABA受体拮抗剂进行干预后发现,仅在白天可引起高血糖症,提示从视交叉上核传递到室旁核的GABAergic神经元信号的昼夜改变会引起肝脏葡萄糖生成的相应改变[6]。视交叉上核的生物钟可受到由食欲素神经元产生的促觉醒信号的重置,而食欲素神经元的激活很大程度上又受到生理节奏的控制,提示两者之间存在互惠关系[7]。
1.3.2食欲素、胰岛素、瘦素和血糖调节的关系 在胰岛素对肝脏葡萄糖生成的抑制效应中,下丘脑信号作用是必须的。下丘脑-肝脏通路的任何改变都可能会造成糖尿病高血糖症。外周胰岛素抵抗相关的高血糖症抑制下丘脑食欲素的表达,而食欲素有助于维持下丘脑胰岛素敏感性,食欲素表达受到抑制之后便产生了下丘脑和外周胰岛素抵抗的恶性循环(图2)[8]。正常情况下,胰岛素在下丘脑的作用可促进胰岛素的降糖效应,特别是通过抑制肝脏葡萄糖生成。这个过程由下丘脑ATP敏感性钾通道(KATP)通道介导的。另外,下丘脑蛋白激酶C(可能是蛋白激酶Cδ亚型)的活化对于激活下丘脑KATP,从而降低肝脏葡萄糖生成是非常必要的。瘦素也可通过信号转导与转录激活因子3通路调节肝脏葡萄糖生成。另一方面,年龄或食源性胰岛素抵抗相关的高血糖症的患者,其下丘脑食欲素的表达减少。反过来,食欲素的缺乏可导致下丘脑胰岛素受体、瘦素受体以及食欲素受体信号中断(具体机制不明)。结果就是调节能量(葡萄糖)代谢的下丘脑/肝脏通路受损,从而进一步加剧外周组织的胰岛素抵抗。

LRb:瘦素受体;OXR:食欲素受体;IR:胰岛素抵抗;PI3K:磷脂酰肌醇3-激酶;Akt:蛋白激酶B;Stat3:信号转导与转录激活因子3;PKC:下丘脑蛋白激酶C
图2食欲素、瘦素、胰岛素循环轴与血糖调节的关系
1.3.3食欲素影响胰岛素、瘦素敏感性的机制 虽然食欲素影响胰岛素、瘦素敏感性的确切机制仍不明确,但是很可能在食欲素缺乏的情况下(例如前食欲素原基因消融或者高血糖症),下丘脑蛋白激酶B磷酸化水平会随着年龄增长而增高,这种长期的磷酸化作用破坏了胰岛素在下丘脑的作用,从而影响外周胰岛素敏感性。
在正常饮食条件下,食欲素基因剔除的小鼠出现随年龄增长呈进行性发展的糖耐量受损和胰岛素抵抗,观察对象包括非肥胖的雄性小鼠和轻度肥胖的雌性小鼠。9月龄的食欲素基因剔除的小鼠,在正常饮食条件下,通过蛋白激酶B通路的胰岛素信号在骨骼肌和肝脏中断。2~6月龄的食欲素基因剔除的小鼠,下丘脑蛋白激酶B(Ser473)磷酸化水平出现异常增加,且胰岛素的刺激不能提升蛋白激酶B磷酸化水平,而在9月龄的食欲素基因剔除小鼠中也观察到胰岛素诱导的胰岛素受体酪氨酸磷酸化减少,提示食欲素是维持下丘脑和外周胰岛素敏感性所必须的,尤其随着年龄的增长更是如此。运用示踪稀释法结合胰岛素钳夹技术的研究证实,下丘脑蛋白激酶C的激活对于降低肝脏葡萄糖生成是非常重要且必须的,主要通过下丘脑磺脲类药物受体1/Kir6.2 KATP的激活/磷酸化,而食欲素A可强烈激活CHO细胞上的蛋白激酶Cδ,稳定表达人OXR1[9]。因此,食欲素可能是通过促进KATP的激活来增强胰岛素在下丘脑的作用,从而抑制肝脏葡萄糖生成。
近几年的研究证实,在啮齿动物中食欲素可强烈抑制食源性肥胖以及后续出现的胰岛素抵抗。食欲素过表达的转基因小鼠可通过增加能量支出、减少能量消耗而预防高脂饮食诱导的肥胖、胰岛素不敏感、高胰岛素血症和高瘦素血症。将食欲素过表达的OXR1和OXR2基因敲除小鼠进行分子遗传分析发现OXR2主要介导了这种抗肥胖效应和改善胰岛素敏感性的效应。持续应用一种OXR2选择性兴奋剂食欲素B([Ala11,d-Leu15]-Orexin B)进行中枢干预可预防食源性肥胖。另外,食欲过表达的转基因小鼠表现为对外源性输注的瘦素的敏感性增加,食欲素过表达或OXR2兴奋剂均不影响瘦素缺乏的肥胖ob/ob小鼠的体质量增加,说明食欲素经OXR2的作用与瘦素敏感性状态相关[10]。需要注意的是,单独的OXR1缺乏可预防高脂饮食诱导的高血糖症和高胰岛素血症,但无法防止肥胖,而食欲素过表达可预防OXR1和OXR2基因敲除的小鼠在高脂饮食的情况下发生肥胖。这些结果提示,内源性食欲素-OXR1信号可能介导了高脂饮食对糖代谢和肥胖的有害作用。事实上,已经有报道一种OXR1拮抗剂SB334867A可能成为抗肥胖和糖尿病的因子,因为它能减少肥胖ob/ob小鼠的摄食量,降低体质量、空腹血糖和血浆胰岛素水平[11]。而食欲素-OXR2信号的增加可防止食源性肥胖,提高瘦素敏感性。
因此,食欲素可通过增加下丘脑胰岛素和(或)瘦素信号来预防年龄相关及饮食诱导的胰岛素抵抗,OXR可作为未来治疗2型糖尿病患者高血糖症的重要靶点。
2 食欲素与能量代谢调节
关于食欲素对能量代谢的调节机制,目前仍不明确。下丘脑食欲素诱导的能量代谢的增加并不仅仅是由于觉醒和身体活动的增加。事实上,食欲素A会引起氧耗增加和体温上升,即使麻醉的大鼠也是如此,小鼠脑室内注射食欲素A能在不改变摄食和体育活动的情况下增加代谢率。另外,食欲素神经元缺失的小鼠表现为能量消耗减少,并且这一改变与睡眠/觉醒、运动和进食无关。
已知线粒体解偶联蛋白1(uncoupling protein 1,UCP1)与非寒战产热有关,并且主要受交感神经系统支配,其原因是β去甲肾上腺素能递质直接支配棕脂肪组织进行调控[12]。这种产热过程在机体能量代谢平衡中非常重要。食欲素A可激活交感神经系统,食欲素基因剔除的小鼠表现为较低的交感缩血管纤维紧张性[13]。然而,长期缓慢室旁核内注射食欲素A并不改变大鼠棕脂肪组织中UCP1的水平[14]。大鼠第三脑室内注射食欲素A后也观察到了类似的结果。此外,低脂饮食的情况下,食欲素过表达的转基因小鼠的棕脂肪组织中UCP1 mRNA的水平与对照组小鼠比较差异无统计学意义[15]。因此,食欲素可能不是通过棕脂肪组织中的UCP1来调控能量代谢的。
与UCP1不同,UCP2在全身广泛表达,包括中枢神经系统,增加UCP2引发的线粒体解耦联可使活性氧类生成减少,产生神经保护效应。UCP2也能增加海马细胞中线粒体的数量。外侧下丘脑区表达的食欲素A和食欲素B为热能的产生提供了一个解剖学上受限的位点[16]。食欲素神经元UCP2过表达的转基因小鼠(orexin/UCP2-Tg mice)下丘脑温度升高,伴随身体核心温度降低。但是,也有一些报道提示与之矛盾的结果,因此食欲素是如何通过UCP2调节能量代谢的机制仍有待进一步研究。
3 食欲素在外周组织的作用
食欲素大部分是在下丘脑发挥其功能,在神经系统以外的作用目前仍存在争议,食欲素及其受体存在于广泛的组织中,包括肠道、胰腺、肾脏、肾上腺、脂肪组织和生殖系统。
研究证实,随着链脲佐菌素诱导的糖尿病的发生,大鼠胰岛细胞中OXR1的表达增加,糖尿病病程越长,胰岛细胞中OXR1的表达越强烈,并且与胰高血糖素共定位[17]。另外,免疫组织化学发现在胰岛有被剪切的胱天蛋白酶3片段与OXR1共定位。食欲素基因剔除的小鼠中OXR1和胱天蛋白酶3的表达显著降低,说明在胰腺组织中食欲素可能是通过OXR1介导促进链脲佐菌素诱导的糖尿病的发展和β细胞凋亡[18-19]。但是,关于食欲素在外周组织中的作用目前仍然知之甚少,有待进一步研究。
4 小结与展望
食欲素参与调控血糖平衡,调节胰岛素/瘦素敏感性,食欲素可有效预防年龄相关或高脂饮食诱导的外周胰岛素抵抗的进行性发展。食欲素-OXR2信号可通过增加瘦素敏感性而预防食源性肥胖和胰岛素抵抗。随着年龄增长,海马、下丘脑、脑桥等组织中OXR2 mRNA水平著显下降,另外,前食欲素原基因表达在肥胖的ob/ob和db/db小鼠出现下调,并且这种下调现象是由高血糖导致的。这些现象都提示,胰岛素敏感性下降所导致的高血糖症造成下丘脑食欲素的表达降低,从而加剧了外周胰岛素抵抗。因此,控制下丘脑胰岛素/瘦素信号的食欲素受体可作为2型糖尿病高血糖症治疗的新靶点。
[1] Burdakov D,González JA.Physiological functions of glucose-inhibited neurones[J].Acta Physiol(Oxf),2009,195(1):71-78.
[2] Marino JS,Xu Y,Hill JW.Central insulin and leptin-mediated autonomic control of glucose homeostasis[J].Trends Endocrinol Metab,2011,22(7):275-285.
[3] Shen J,Tanida M,Yao JF,etal.Biphasic effects of orexin-A on autonomic nerve activity and lipolysis[J].Neurosci Lett,2008,444(2):166-171.
[4] Shiuchi T,Haque MS,Okamoto S,etal.Hypothalamic orexin stimulates feeding-associated glucose utilization in skeletal muscle via sympathetic nervous system[J].Cell Metab,2009,10(6):466-480.
[5] Ramadori G,Fujikawa T,Anderson J,etal.SIRT1 deacetylase in SF1 neuronsprotects against metabolic imbalance[J].Cell Metab,2011,14(3):301-312.
[6] Kalsbeek A,Yi CX,La Fleur SE,etal.The hypothalamic clock and its control of glucose homeostasis[J].Trends Endocrinol Metab,2010,21(7):402-410.
[7] Marston OJ,Williams RH,Canal MM,etal.Circadian and dark-pulse activation of orexin/hypocretin neurons[J].Mol Brain,2008,1:19.
[8] Ross R,Wang PY,Chari M,etal.Hypothalamic protein kinase C regulates glucose production[J].Diabetes,2008,57(8):2061-2065.
[9] Johansson L,Ekholm ME,Kukkonen JP.Multiple phospholipase activation by OX1 orexin/hypocretin receptors[J].Cell Mol Life Sci,2008,65(12):1948-1956.
[10] Funato H,Tsai AL,Willie JT,etal.Enhanced orexin receptor-2 signaling prevents diet-induced obesity and improves leptin sensitivity[J].Cell Metab,2009,9(1):64-76.
[11] Haynes AC,Chapman H,Taylor C,etal.Anorectic,thermogenic and anti-obesity activity of a selective orexin-1 receptor antagonist in ob/ob mice[J].Regul Pept,2002,104(1/3):153-159.
[12] Golozoubova V,Hohtola E,Matthias A,etal.Only UCP1 can mediate adaptive nonshivering thermogenesis in the cold[J].FASEB J,2001,15(11):2048-2050.
[13] Kayaba Y,Nakamura A,Kasuya Y,etal.Attenuated defense response and low basal blood pressure in orexin knockout mice[J].Am J Physiol Regul Integr Comp Physiol,2003,285(3):R581-R593.
[14] Russell SH,Small CJ,Sunter D,etal.Chronic intraparaventricular nuclear administration of orexin A in male rats does not alter thyroid axis or uncoupling protein-1 in brown adipose tissue[J].Regul Pept,2002,104(1/3):61-68.
[15] Conti B,Sanchez-Alavez M,Winsky-Sommerer R,etal.Transgenic mice with a reduced core body temperature have an increased life span[J].Science,2006,314(5800):825-828.
[16] Adeghate E,Fernandez-Cabezudo M,Hameed R,etal.Orexin-1 receptor co-localizes with pancreatic hormones in islet cells and modulates the outcome of streptozotocin-induced diabetes mellitus[J].PLoS One,2010,5(1):e8587.
[17] Sakurai T,Mieda M.Connectomics of rexinproducing neurons:interface of systems of emotion,energy homeostasis and arousal[J].Trends Pharmacol Sci,2011,32(8):451-462.
[18] Carter ME,Borg JS,de Lecea L.The brain hypocretins and their receptors:mediators of allostatic arousal[J].Curr Opin Pharmacol,2009,9(1):39-45.
[19] Yi CX,Serlie MJ,Ackermans MT,etal.A major role for perifornical orexin neurons in the control of glucose metabolism in rats[J].Diabetes,2009,58(9):1998-2005.
