生物信息学在药物研究和开发中的应用
2014-03-23王可鉴
王可鉴,贺 林,杨 仑,2,3
(1.上海交通大学Bio-X研究院,上海200030;2.Glaxosmithkline,Systematic Drug Repositioning,Philadelphia 19406,USA;3.School of Pharmacy,Medical School,University of South Florida,Tampa 33620,USA)
生物信息学在药物研究和开发中的应用
王可鉴1,贺 林1,杨 仑1,2,3
(1.上海交通大学Bio-X研究院,上海200030;2.Glaxosmithkline,Systematic Drug Repositioning,Philadelphia 19406,USA;3.School of Pharmacy,Medical School,University of South Florida,Tampa 33620,USA)
生物信息学作为一门综合计算机科学、信息技术和数学理论开发新的算法和统计方法的学科,对生物实验数据进行分析从而确定数据中所隐含的生物学意义,并开发新的数据分析工具以实现对各种信息的获取和管理的交叉学科。其主要优势在于以低成本和高通量的方式对大量生物学和医学数据进行管理和分析,侧重于从中进一步挖掘与药物疗效、作用机制和副作用等相关的有价值的信息,为药物研究提供参考和指导。基于低通量药理或毒理学实验的传统新药研发流程具有周期长、成本高和失败率高的局限性。结合其成功运用药物生物信息学进行新药研发和旧药新用的经验,本综述介绍了药物生物信息学在新药研发中的新进展,表明在我国建设药物生物信息学平台的重要性和必要性。
新药;生物信息学;药物不良反应
DOl:10.3867/j.issn.1000-3002.2014.01.018
新药研发过程中需要解决的一个十分重要却又异常复杂的问题是:药物以何种方式影响了哪些通路以及通路中的哪些基因,进而如何产生药效或毒性。通常情况下,药理学、毒理学或分子生物学实验是主要的探究方法。然而,考虑到这些方法或平台所需的资金和人力往往难以达到最佳的投入产出比,生物信息学方法则能体现出其特有的优势。例如在计算机辅助下,通过对基因组学和其他组学数据进行分析,能够以较小资金投入实现对“药物-基因”关系的高通量筛选,从而降低了科研风险。应当说,在当前新药研发的各种主流策略中,生物信息学越来越受到重视,分析高通量组学数据的生物信息学方法发挥着重要的作用。
1 药物生物信息学研究的基本原理
生物信息学在生命科学研究中,是一门综合计算机科学、信息技术和数学理论开发新的算法和统计方法的交叉学科,对生物实验数据进行分析从而确定数据中所隐含的生物学意义,并开发新的数据分析工具以实现对各种信息的获取和管理[1]。生物信息学是随着人类基因组计划的启动而兴起的,其研究范围涵盖了生物学数据的整理、存档、显示、计算和模拟,基因遗传和物理图谱的处理,核苷酸和氨基酸序列分析,新基因的发现和蛋白质结构的预测等方面。而药物生物信息学作为生物信息学的一个重要独立分支学科,则更侧重于处理与药物直接相关的知识和数据,如旧药新用、不良反应、药物作用机制、药物相互作用和电子病历等信息,并在临床转化和新药研发中发挥着重要作用[2]。当前,药物相关的研发受益于生物技术和计算机技术的进步,并不断产生着大量的与药物作用和药物反应相关的高通量数据,例如基因组学[3]、转录组学、蛋白质组学和代谢组学等等[4]。因此,当今药物研发在信息处理层面上的主要瓶颈不再是数据的匮乏,而是数据过剩带来的信息解读的不充分。也就是说,药理学和毒理学研究的发展和进步不仅要依靠更新更快的硬件平台来产生数据,也要依靠更有效更可靠的各种算法、软件和工具来对大量抽象的实验数据进行清晰而准确的分析。而药物生物信息学就是挖掘和利用实验数据中的信息,从众多的数值、文本和序列中去伪存真,发现统一的、系统性的药理或临床规律,并将这些规律总结成可读可视可用的格式(如图表、公式和软件等)以供后续的研究参考和使用,从而促进高效和安全的药物研发。可以预见,随着生命科学的研究方向不断向系统生物学的思维靠拢,药物研发也将越来越多地遵循系统药理和毒理学的研究思路,其中药物生物信息学必将发挥越来越重要的作用[5]。
2 药物生物信息学的新发展
从理论上讲,药理学、毒理学以及其他与药物的结构、作用和反应相关的研究都与生物信息学的研究范畴存在交叉。所以,生物信息学方法在药物相关的研究中也得到了越来越深入和广泛的应用[6]。并且,伴随着基因组学的进步和药物研发的升级,逐渐演化出了药物生物信息学这一门应用性、实践性很强的学科分支。它综合应用生命科学、数学、计算机科学等多学科的理论和方法,对伴随基因组计划产生的海量生物信息进行整理和分析,然后将结果应用于药物的设计、优化和开发,以实现更加合理的药物设计与使用作为最终目的。
在新药研发的流程中,几乎每一个主要步骤都有生物信息学发挥的重要作用[7](图1)。①先导化合物筛选:药物研发的本质就是发现具有特定生物学和药理学活性的化合物分子。而这类分子的前身就是通常所说的“先导化合物”[8],以往需要在海量的化合物分子中通过生物化学方法进行大规模的筛选,其成本极其高昂。在生物信息学定量构效关系模型(QSAR)的帮助下,可以总结出某些规律,指示具有特定化学结构的分子更倾向于具有特定的药理作用。比如运用药物分子与靶蛋白模拟对接法[9]和以药效团作为提问结构的三维结构搜索法[10]能够从数据库中搜索所需的先导化合物。当直接从数据库中搜索不到所需的化合物分子时,可运用化合物设计方面的生物信息学工具重新对先导化合物分子进行构建[11],这些方法从多个角度提高了化合物筛选的效率并降低了成本,业已成为现代药物研发的常规必要手段。② 靶蛋白发现:药靶发现历来是药物开发的先决条件,人类基因组测序的不断完善为药物研究提供了大量的潜在靶基因,但如何在众多的基因和序列中找到真正有效的药物靶标是一个制药业必须面对的严峻考验[12]。应用生物信息学方法可以对已知有效的靶基因进行量化分析,比如总结其在核苷酸和氨基酸序列方面的特性,并将其他一系列人类基因与经典靶基因进行基因结构上的同源性对比,从而快速确定新基因是否有潜力成为新药靶[13],这样就可以避免盲目而草率地进行相对昂贵的实验验证。而最近各种新兴的脱靶(o ff-target)挖掘技术的产生[14-22],又为新靶点的发现提供新的候选。比如,通过计算机模拟可实现数百个化合物分子和数百个人类蛋白之间的模拟对接,得到了一个大规模的“药物-蛋白互作组”(CPI)矩阵,从中挖掘出一系列与旧药新用或不良反应相关的药物脱靶现象[19,22-26]。如果要通过传统的化学方法分析如此数量的药物和药靶之间的未知关系,将需要投入大量的人力物力且必须经过一个较长的研究周期。③药物作用机制:在确定了药物分子的化学结构和靶蛋白之后,通常还需要进一步研究药物与机体不同组织和细胞之间相互作用的结果,从而确定药物可通过何种方式对潜在适应证产生疗效以及可产生的副作用。在传统的药物研发中,这一阶段的研究主要基于动物模型实验。由于动物培养和药物处理通常所需的时间很长且成本极高,所以数据规模有限,难以进行不同药物之间横向的对比,影响了研究结论的系统性和可靠性。通过结合大规模的微阵列和创新的生物信息学分析方法(详见本文第3部分),可以在基因组表达层面上比较不同药物在作用机制上的相似性,在很大程度上促进了药物机制的研究。④临床统计分析:经过不断优化和测试的化合物究竟是否成为一个成功的药物,其最终的评价标准还在于临床应用的效果,在这一方面生物信息学的参与也极具重要性[24]。众所周知,动辄涉及数千患者的临床试验是药物开发流程中成本最高的步骤,如果由于患者样本的选择不合理而引起统计学上的偏差,则无异于拿患者宝贵的生命和巨大的资源投入来冒险。而使用生物信息学工具在基因组水平对临床受试的患者群体进行有效的划分和甄别,可以大大提高新药开发的效率并节省大量的时间和经费。比如,美国斯坦福大学的Tatonetti等[25-26]收集了大量的临床试验数据和电子医疗记录并进行整理,在分析中有效消除了其中性别年龄等协变量的干扰。通过观察药物在大的人群样本中倾向于引起那些特定的药物反应,可以从中发现未知的新药靶、药物适应证和药物联用。
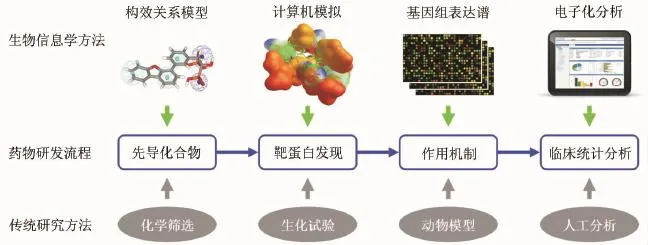
图1 药物生物信息学方法在药物研发中的应用.
随着生物技术的进步和数据生成的爆炸式增长,上述的越来越多数据都将需要生物信息学加以组织和分析[27],从而实现对药物与适应证关系的总结、药物不良反应事件的监测、药物未知作用机制研究以及旧药新用(药物重定位)[28-30]和不良反应的预测等。大型医疗信息数据库是生物信息学在临床药理研究中最早的应用方向,即利用计算机把服药者分散的医疗信息汇总并重新组织,从而建立可供记录和查询大型数据库。迄今,已经有一系列数据库成功地应用于相关研究。利用信息学在标准化评价上的优势,研究者得以对服药者对药物的反应进行客观的因果关系分析并以概率定量的形式全面准确评价所有可能的影响因素[31]。在数据库的高效框架下,生物信息学通过对药物数据的储存、处理、分析来“提炼”包含在海量数据中的相关知识,及其生物学和药理学意义,从而为破解药物作用的内在机制提供线索。研究机制的最终目的是实现准确的预测,即最大限度地避免不良反应导致的人身伤害和经济损失,或者最大限度地抓住对旧药物进行重新定位的机会。单纯依靠分子生物学实验的研究方法通常耗资大,周期长;而对生物信息学工具[32]加以充分利用,使其与生物学实验验证相配合,则能够取长补短、发挥合力。研究表明,现有的预测工具虽然在预测效率方面仍然有提高的空间,但至少能够在一定程度上为化学、药理、毒理学家和制药公司等提供决策支持[33-34]。这说明药物生物信息学方法作为一种与传统生物学实验和临床试验方法互补的新研究途径,有望为药物开发开拓出一条新路。
3 药物生物信息学在新药研发中的应用:基于表达谱的药物研究
虽然许多研究都致力于解释药物分子在上游结合靶蛋白之后,间接地影响了哪些下游基因,以及这些下游基因之间又发生了怎样的相互作用和形成了怎样的网络[25,35],但是想要通过可视化的图表和可定量的数据来描述每一个受到药物影响的下游基因却是件十分艰巨的任务。一方面,生物体内基因与基因之间的关系是一个有机的整体,当上游的药物作用影响到了第一批下游基因后,受影响的基因又会继续调控第二批、第三批基因,而且这种波浪式扩散中往往还包含着正反馈[36]和负反馈[37]效应,因此任何一个药物对体内基因的作用都可谓“牵一发而动全身”。另一方面,对于如此复杂的药物作用,如果单纯依靠分子生物学实验来进行逐个基因的验证无疑将耗费大量的资源和时间。按照生物学发展的现有条件来说,要同时确定一大批基因的表达变化的方式和程度,可行性和可靠性最高的方案就是通过基因芯片平台对整个基因组的表达水平进行高通量的检测。
由于基因芯片技术的普及,在生物学领域的各大公共数据库中已经公开了大量药物相关的基因组表达数据。其中对基因芯片比较典型的应用方式是使用各种药物处理人类或动物组织的细胞[38],通过比较药物处理前后细胞内基因表达谱可以得到差异表达基因(differentially expressed gene),用来代表药物的药效特征信号,或者称为基因组中的特征基因。假设药物A引起一部分基因表达上调和另一部分基因表达下调;同时某个治疗另一种适应证与A不同的药物B处理细胞后,A引起上调(或下调)的基因在B处理的细胞中也普遍倾向于表达升高(或降低)。这种情况表明A和B调控的基因具有高度正相关的表达变化,也就是说两种药物影响下游基因的方式高度相似,则A和B很可能在作用机制方面存在某种未知的重叠或关联。从类似A和B这样的“药物-药物”配对中寻找新的适应证定位即检验A是否能够治疗B的适应证,或B治疗A的适应证显然比随机的筛选或经验型地摸索具有更高的效率和成功率。基于以上逻辑的一系列研究都取得了令人信服的结果[39],足见通过计算药物之间在基因组表达谱上的相关度来对比它们在作用机制上的相似性是一条完全可行的研究道路[40]。然而,由于不同的基因芯片、不同的实验设计甚至不同的实验人员操作习惯都能对基因芯片实验的结果产生一定程度的影响,因此相互独立的小规模实验所产出的药物特征基因表达谱往往不具备足够的相互之间的可比性,也就难以进行统一的数据分析来实现大规模的新药研发。
2006年,美国Broad研究所的Lamb等[41-42]在美国《Science》上发表了其建立的大规模药物处理人类细胞的数据库以及相关研究成果。在这个被被命名为“联系图”(connectivity map,CMap)的药物基因组学研究项目中,研究人员首先使用1309种药物对6100个人类细胞系进行了处理(图2)。处理之后的细胞相对于未经处理的对照细胞必定产生了某种程度的基因表达变化。其后,研究人员利用Affymetrix公司的微阵列技术平台对所有细胞中的20 000余个基因进行了定量检验,从而明确地测量出每个细胞系在特定药物处理后有哪些基因发生了表达水平的上调或下调,这些信息即构成了该药物特有的“基因组表达谱”。最后,通过对比不同药物之间在基因组表达谱上的相关程度即可判断药物在调控下游基因的方式上的相似程度,也就间接反映了不同药物是否影响了相近的或部分重叠的下游基因。例如,如果某个治疗糖尿病的药物在CMap中的基因组表达谱体现出与多个抗癌药物的显著正向相关性,则预示着该糖尿病药物很可能调控了癌症相关基因并具有潜在的抗癌作用。
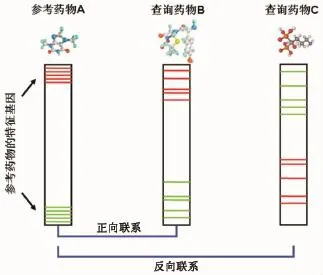
图2 联系图的设计原理.
为了定量衡量一对药物表达谱之间正向或负向的相关程度,CMap根据特定的计算方法能够给出一个介于-1和+1之间的“联系分值”,用以定量衡量各药物表达谱之间的相似程度。首先在药物A的基因组中选择表达差异最大的一部分基因探针(比如上调和下调各500个,合计1000个探针)作为其“特征基因”。随后,将药物B的基因组中所有22 283个基因探针与药物的特征基因进行比对。如果A的上调特征基因(下调同理)在B中更多地倾向于过表达,则代表两药在基因调控方面的相似程度越高。该程度可以量化为以下分值:
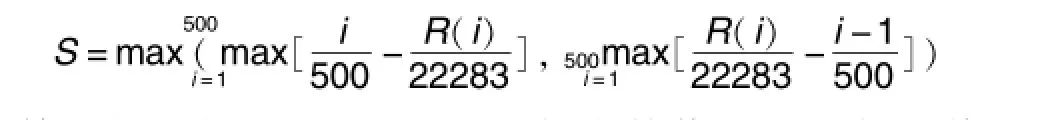
第i个(i为1~500之间的某个数值)在B表达谱中的上调排名(介于1~22 283之间)。基于此计算公式,可以实现不同药物之间高效率的基因组表达定量比对。在过往以CMap为基础的众多研究中,主要的思路就是以某个药物的基因组表达谱为参照,将其在整个CMap数据中进行“查询”(即将所有的CMap基因组表达谱与之进行比对),最终正向联系分值最高(最接近于+1)的一部分药物便被认为可能与参照药物之间存在下游基因调控和临床药物反应层面上的潜在关联。
对于每个细胞样本来说,其中一部分加入特定药物而成为“处理组”,而另一部分未作任何处理便作为“对照组”。处理组相对于对照组细胞来说,各个基因的表达水平应当都发生了某种程度的变化,而整个基因组的整体变化水平就是该处理药物特有的基因组表达谱。当把某个药物作为参照标准时,其基因组表达谱中上调和下调倍数最高的一部分基因就被选为该药物的特征基因。如果要查询某个其他药物与参照药物在基因组表达谱上的相似程度,只需计算参照药物的特征基因在查询药物的表达谱中的上调和下调水平。如果参照药物的上调(或下调)基因在查询药物处理的细胞中也普遍倾向于上调(或下调),则两者的表达谱呈正向相关;反之,如果参照药物的上调(或下调)基因却在查询药物处理的细胞中呈现出普遍下调(或上调)的趋势,则两者为反向相关。
自从发表以来,各方基于CMap数据的研究主要集中在旧药新用方面,而且取得了一系列不同程度的进展[43-44]。还有研究以改进CMap基本分析方法为切入点,通过一定的预处理程序提高CMap数据的质量[45-46],再用升级后的CMap发掘出之前被埋没在数据噪声中的信号,从而展示了数据预处理对药物研发的重要性。在此对若干最具代表性的应用性研究加以详细介绍。
发现具有抗癌活性的新药物,是生命科学的热点领域之一。而如何以低投入和高效率的方式发现抗癌新药始终是一个难点问题。意大利Tele Thon研究所的Isacchi研究组广泛分析了所有1309个CMap药物之间的表达谱相似度,并从中发现了一系列相互紧密联系并具有相似疗效的的“药物群”[47]。比如,2DOG(2-deoxy-D-glucose)分子已知能够引发细胞自噬作用,一种与癌症、感染和神经退行性疾病广泛相关的生物学过程。而在药物群中,2DOG和降血压药物法舒地尔体现出了显著的强联系,表明法舒地尔具有潜在的引发细胞自噬的能力。为了验证这一CMap生成的线索,分别以人类成纤维细胞和HeLa细胞为模型,该研究组检测了法舒地尔对细胞自噬作用标志物LC3-Ⅱ水平变化的影响。免疫印迹和抗体免疫染色的双重证据都证实,法舒地尔引发了明显的细胞自噬,从而通过低成本和高通量的数据分析精准发现了法舒地尔的潜在抗癌疗效。
美国斯坦福大学的Butte研究组基于CMap方法进行了成功的老药新用研究,并在《Nature Transl Med》上发表了2篇系列文章。该研究组首先从美国国立卫生研究院(NIH)的Gene Expression Omnibus数据库中收集了大量数据,最终提取出100个疾病模型和164个化合物小分子的基因组表达谱信息。通过CMap的分析方法,可以计算药物分子与疾病模型的反向联系。比如疾病模型中上调(或下调)的基因在药物处理中普遍下调或(上调),则很可能提示了未知的“药物-适应证”关系。比如,抗惊厥药托吡酯被发现可能具有治疗炎症性肠病的作用[48],而在硝基苯磺酸引发的炎症性肠病动物模型中,口服托吡酯显著减轻了总体病理特征和受感染结肠组织的为损伤。同时,抗溃疡药物西咪替丁(甲氰咪胍)具有潜在的抗癌疗效的老药新用假设也在动物疾病模型中得到了有效验证[49]。
美国爱荷华大学的Adams研究组通过反向比对的方法,提出了针对肌肉萎缩这一常见疑难疾病的新疗法。该研究组首先从人类和大鼠的肌肉样本中定位出63个与肌肉萎缩显著相关的m RNA片段,用以表征相关的体内病理环境。随后,通过将疾病特征m RNA与CMap药物的基因组表达模式进行对比,寻找到了包括熊果酸在内的一部分与疾病样本显著相反的药物。该研究组认为,那些与病理样本相反的药物有可能扭转肌肉萎缩的体内环境,从而实现对疾病的治疗。最后,动物模型实验证明熊果酸确实刺激了与肌肉生长相关的信号通路,并且有效提升了动物体内的肌肉重量。相关结果发表在《Cell Metabolism》上,为肌肉萎缩的治疗提供全新的临床研究方向。
在积极应用药物基因组表达数据同时,研究者们也注意到以CMap为代表的大规模表达数据在数据质量上可能存在着一些不容忽视的问题。其中最主要的问题就是大规模生物学实验中常见的“批次效应”,即数据信号的主要决定因素并非样本本身的生物学特征,而是样本在获取、培养或测量过程中周围环境的微小差异。具体到CMap数据中,由于全部6100个细胞样本是在302个不同批次中培养出来,研究人员发现,同一批次不同药物处理的样本之间的表达相似度甚至高于同一药物不同批次的细胞样本(即细胞培养条件的噪声掩盖了药物作用的信号)。为了克服这一不足,生物信息学家们从不同角度提出了多个解决方案。研究者发现,对于同一药物处理的不同批次细胞来说,其差异主要体现在细胞条件而非药物作用,所以其表达谱的差异基本能够反映细胞批次间的差异。通过对此差异进行定量计算,可以对整个CMap进行系统性的校正。结果表明,校正后的CMap表达谱更真实地反映了药物作用的特性,并成功地被用来研究药物药靶的表达[50]。美国葛兰素史克制药公司的科学家也发现,通过对CMap联系分值的计算方法进行改进,以基因表达向量之间的“余弦值”作为衡量相似度的标准,可以有效突出药物作用在表达谱中的信号,并借此成功地预测了旧药新用的机会[51]。可以预见,通过不断的优化实验设计和生物信息学方法,CMap及其他大规模基因组表达数据受到批次效应的影响将越来越弱,并越来越有力地促进基于生物信息学的药物研发。
4 展望
综合以上众多的理论性和应用性研究,可以发现在系统生物学理论的指导下,基于药物生物信息学方法对药物相关的高通量数据进行分析和挖掘,能够在传统的经验性临床摸索和低通量实验之外,建立一套更高效的新药研发的方法论。较之实验药理、毒理学和临床医学研究,药物生物信息学方法在研究成本具有优势,研发周期也相对更短,特别是在临床样本收集和动物模型建立十分困难的研究“初级阶段”,可采用系统生物学理论和生物信息学方法摸清大致脉络,从而实现在前期较少经费的基础上,为后续实验提供可行可靠的假说,也就为后续长期大额经费的投入指明了正确的方向。因此,建立并完善我国药物生物信息学平台,对于研发适合我国国情的旧药新用(药物新组合),或预测并预防药物不良反应具有重大意义,是我国新药创制不可缺少的重大平台。
但同时我们也必须认识到,单纯依靠药物生物信息学分析并不能直接得到新的药物和疗法,而是最终要落实到生物化学和临床医学实验当中。单纯依靠实验而忽视了在计算层面上对信息和数据的深入分析,则难以有效地降低药物研发成本和提高研发效率。因此,未来药物生物信息学乃至整个制药工业的发展方向必将是信息学、药理、毒理学和医学的高度协作。
[1] Wada A.Bioinformatics-the necessity of the quest for″first principles″in life[J].Bioinformatics,2000,16(8):663-664.
[2] Altman RB.Introduction to translational bioinformatics collection[J].PLoS Comput Biol,2012,8(12):e1002796.
[3] Xiao L,Zhang J,Sirois P,He N,Li K.A new strategy for next generation sequencing:merging the Sanger′s method and the sequencing by synthesis through rep lacing extension[J].J Biomed Nanotechnol,2011,7(4):568-571.
[4] Schneider MV,Orchard S.Omics technologies,data and bioinformatics principles[J].Methods Mol Biol,2011,719:3-30.
[5] Kumar S,Dudley J.Bioinformatics software for biologists in the genomics era[J].Bioinformatics,2007,23(14):1713-1717.
[6] Whittaker PA.What is the relevance of bioinformatics to pharmacology?[J].Trends Pharmacol Sci,2003,24(8):434-439.
[7] Terstappen GC,Reggiani A.In silico research in drug discovery[J].Trends Pharmacol Sci,2001,22(1):23-26.
[8] Lewell XQ,Jones AC,Bruce CL,Harper G,Jones MM,McLay IM,et al.Drug rings database with web interface.A tool for identifying alternative chemical rings in lead discovery programs[J].J Med Chem,2003,46(15):3257-3274.
[9] Chakraborty B,Roy AS,Dasgupta S,Basu S.Magnetic field effect corroborated with docking study to explore photoinduced electron transfer in drug-protein interaction[J].J Phys Chem A,2010,114(51):13313-13325.
[10] Huang D,Lüthi U,Kolb P,Edler K,Cecchini M,Audetat S,et al.Discovery of cell-permeable nonpeptide inhibitors of beta-secretase by highthroughput docking and continuum electrostatics calculations[J].J Med Chem,2005,48(16):5108-5111.
[11] Yang J,Zhan CY,Dong XC,Yang K,Wang FX.Interaction of human fibrinogen receptor(GPⅡb-Ⅲa)with decorsin[J].Acta Pharmacol Sin,2004,25(8):1096-1104.
[12] Berry S.Drug discovery in the wake of genomics[J].Trends Biotechnol,2001,19(7):239-240.
[13] FerrariS,Losasso V,CostiMP.Sequence-based identification of specific drug target regions in the thymidylate synthase enzyme family[J].Chem Med Chem,2008,3(3):392-401.
[14] Xie L,Wang J,Bourne PE.In silico elucidation of the molecular mechanism defining the adverse effect of selective estrogen receptor modulators[J].PLoS Com put Biol,2007,3(11):e217.
[15] Campillos M,Kuhn M,Gavin AC,Jensen LJ,Bork P.Drug target identification using side-effect similarity[J].Science,2008,321(5886):263-266.
[16] Keiser MJ,Setola V,Irwin JJ,Laggner C,Abbas AI,Hufeisen SJ,et a l.Predicting new molecular targets for known drugs[J].Nature,2009,462(7270):175-181.
[17] Yang L,Chen J,He L.Harvesting candidate genes responsible for serious adverse drug reactions from a chemical-protein interactome[J].PLoS Com put Biol,2009,5(7):e1000441.
[18] Luo H,Chen J,Shi L,Mikailov M,Zhu H,Wang K,et al.DRAR-CPI:a server for identifying drug repositioning potential and adverse drug reactions via the chemical-protein interactome[J].Nucleic Acids Res,2011,39(Web Server issue):W492-W 498.
[19] Yang L,Luo H,Chen J,Xing Q,He L.SePreSA:a server for the prediction of populations susceptible to serious adverse drug reactions implementing the methodology of a chemical-protein interactome[J].Nucleic Acids Res,2009,37(Web Server issue):W 406-W412.
[20] Yang L,Wang K,Chen J,Jegga AG,Luo H,Shi L,et al.Exploring off-targets and off-systems for adverse drug reactions via chemical-protein in teractome-clozapine-induced agranulocytosis as a case study[J].PLoS Comput Biol,2011,7(3):e1002016.
[21] Yang L,Chen J,Shi L,Hudock MP,Wang K,He L.Identifying unexpected therapeutic targets via chemical-protein interactome[J].PLoS One,2010,5(3):e9568.
[22] Yang L,W ang KJ,Wang LS,Jegga AG,Qin SY,He G,et al.Chemical-protein interactome and its application in o ff-target identification[J].Interdiscip Sci,2011,3(1):22-30.
[23] Luo H,Chen J,Shi L,Mikailov M,Zhu H,Wang K,et a l.DRAR-CPI:a serve r for identifying drug repositioning potential and adverse drug reactions via the chemical-protein interactome[J].Nucleic Acids Res,2011,39(Web Server issue):W492-W498.
[24] ZvárováJ.IMIA Conference″Statistical Methodology in Bioinformatics and Clinical Trials″[J].Methods In f Med,2006,45(2):137-138.
[25] Tatonetti NP,Denny JC,Murphy SN,Fernald GH,Krishnan G,Castro V,et al.Detecting drug interactions from adverse-event reports:interaction between paroxetine and pravastatin increases blood glucose levels[J].Clin Pharmacol Ther,2011,90(1):133-142.
[26] Tatonetti NP,Liu T,Altman RB.Predicting d rug side-effects by chemical system s biology[J].Genome Biol,2009,10(9):238.
[27] Qu XA,Gud ivada RC,Jegga AG,Neumann EK,Aronow BJ.Inferring novel disease indications for known drugs by semantically linking drug action and disease mechanism relationships[J].BMC Bioinformatics,2009,10(Suppl5):S4.
[28] Zhang YX,Cheng XR,Zhou WX.Drug repositioning:an important application of network pharmacology[J].Chin J Pharmacol Toxicol(中国药理学与毒理学杂志),2012,26(6):779-786.
[29] Yang L,Agarwal P.Systematic drug repositioning based on clinical side-effects[J].PLoS One,2011,6(12):e28025.
[30] Sardana D,Zhu C,Zhang M,Gudivada RC,Yang L,Jegga AG.Drug repositioning for orphan diseases[J].Brief Bioinform,2011,12(4):346-356.
[31] Kramer MS,Leventhal JM,Hutchinson TA,Feinstein AR.An algorithm for the operational assessment of adverse drug reactions.Ⅰ.Background,description,and instructions for use[J].JAMA,1979,242(7):623-632.
[32] Vroling B,Sanders M,Baakman C,Borrmann A,Verhoeven S,Klomp J,et al.GPCRDB:information system for G protein-coup led receptors[J].Nucleic Acids Res,2011,39(Database issue):D309-D319.
[33] von Eichborn J,Murgueitio MS,DunkelM,Koerner S,Bourne PE,Preissner R.PROMISCUOUS:a database fo r network-based drug-repositioning[J].Nucleic Acids Res,2011,39(Database issue):D1060-D1066.
[34] Tan NX,Rao HB,Li ZR,Li XY.Prediction of chemical carcinogenicity by machine learning approaches[J].SAR QSAR Environ Res,2009,20(1-2):27-75.
[35] Yang L,Xu L,He L.A CitationRank algorithm inheriting Google technology designed to highlight genes responsible for serious adverse drug reaction[J].Bioinformatics,2009,25(17):2244-2250.
[36] Veelken H,Leutgeb B,Kulmburg P,Fiebig HH,Mackensen A,Lindemann A.Enhancement of a constitutively active promoter for gene therapy by a positive feed-back transcriptional activator mechanism[J].Int J Mol Med,1998,2(4):423-428.
[37] Lombardi L,Grignani F,Sternas L,Cechova K,Inghirami G,Dalla-Favera R.Mechanism of negative feed-back regulation of c-myc gene expression in B-cells and its inactivation in tum or cells[J].Curr Top Microbiol Immunol,1990,166:293-301.
[38] Zhou M,Liu Z,Zhao Y,Ding Y,Liu H,Xi Y,et al.MicroRNA-125b confers the resistance of breast cancer cells to paclitaxel through suppression of pro-apoptotic Bcl-2 antagonist killer 1(Bak1)expression[J].J Biol Chem,2010,285(28):21496-21507.
[39] Covell DG,Wallqvist A,Huang R,ThankiN,Rabow AA,Lu XJ.Linking tum or cell cytotoxicity to mechanism of d rug action:an integ rated analysis of gene expression,small-molecule screening and structural databases[J].Proteins,2005,59(3):403-433.
[40] Wen Z,Wang Z,Wang S,Ravula R,Yang L,Xu J,et al.Discovery of molecular mechanism s of traditional Chinese medicinal formula Si-Wu-Tang using gene expression microarray and connectivity map[J].PLoS One,2011,6(3):e18278.
[41] Lamb J.The connectivity map:a new tool for biomedical research[J].Nat Rev Cancer,2007,7(1):54-60.
[42] Lamb J,Craw ford ED,Peck D,Modell JW,Blat IC,Wrobel MJ,et al.The connectivity map:using gene-expression signatures to connect small molecules,genes,and disease[J].Science,2006,313(5795):1929-1935.
[43] Wang G,Ye Y,Yang X,Liao H,Zhao C,Liang S.Expression-based in silico screening of candidate therapeutic com pounds for lung adenocarcinoma[J].PLoS One,2011,6(1):e14573.
[44] McArt DG,Zhang SD.Identification of candidate small-molecule therapeutics to cancer by gene-sig-nature perturbation in connectivity mapping[J].PLoS One,2011,6(1):e16382.
[45] Kadota K,Shimizu K.Evaluating methods for ranking differentially expressed genes applied to microArray quality control data[J].BMC Bioinformatics,2011,12:227.
[46] Iskar M,Campillos M,Kuhn M,Jensen LJ,van Noort V,Bork P.Drug-induced regulation of target expression[J].PLoS Comput Biol,2010,6(9):e1000925.
[47] Iorio F,Bosotti R,Scacheri E,Belcastro V,Mithbaokar P,Ferriero R,et al.Discovery of d rug mode of action and drug repositioning from transcriptional responses[J].Proc Natl Acad Sci USA,2010,107(33):14621-14626.
[48] Dudley JT,Sirota M,Shenoy M,Pai RK,Roedder S,Chiang AP,et al.Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease[J].Sci Transl Med,2011,3(96):96 ra76.
[49] Sirota M,Dudley JT,Kim J,Chiang AP,Morgan AA,Sweet-Cordero A,et al.Discovery and preclinical validation of drug indications using compendia of public gene expression data[J].Sci Transl Med,2011,3(96):96ra77.
[50] Chen C,Grennan K,Badner J,Zhang D,Gershon E,Jin L,et al.Removing batch effects in analysis of expression microarray data:an evaluation of six batch adjustment methods[J].PLoS One,2011,6(2):e17238.
[51] Cheng J,Xie Q,Kumar V,Hurle M,Freudenberg JM,Yang L,et al.Evaluation of analytical methods for connectivity map data[J].Pac Symp Biocomput,2013:5-16.
Application of bioinformatics in pharmaceutical research and development
WANG Ke-jian1,HE Lin1,YANG Lung1,2,3
(1.Bio-X Institutes,Shanghai Jiao Tong University,Shanghai 200030,China;2.Glaxosmithkline,Systematic Drug Repositioning,Philadelphia 19406,USA;3.School of Pharmacy,Medical School,University of South Florida,Tam pa 33620,USA)
New drug development is a hot point in life sciences.For a developing country like China,it is always economically and socially important to deliver a series of new drugs or repurposed drugs in order to provide people with safe,effective and affordable therapies.Because of low throughput pharmacology/toxicology assays and clinical trials,traditional drug development pipelines are subject to long cycles,high cost and failure risk.As an interdisciplinary subject,bioinformatics shows specific advantage in management and processing of large-scale biological and medical data,at relatively low cost and high throughput.In drug development,bioinformatics is more often used to discover drug efficacy,mechanisms of action and side effects and to guide research and development.Based on the experience of d rug development and drug repositioning in international pharmaceutical enterprises,the authors proposed the application of bioinformatics to new drug development and illustrated the importance and necessity of bioinformatics platforms in China.
new drugs;bioinformatics;adverse d rug reaction
YANG Lun,E-mail:Lun.Yang@gm ail.com;HE Lin,E-mail:helin@bio-x.cn,Tel:(021)62933338
R318.04
A
1000-3002(2014)01-0118-08
2013-05-24 接受日期:2013-10-18)
(本文编辑:付良青)
王可鉴(1984-),男,博士研究生,主要从事生物信息学研究;杨 仑(1981-),博士,研究员,主要从事药物生物信息学、旧药新用和个性化用药的信息学研究;贺 林(1953-),男,博士,教授,主要从事转化医学研究。
杨 仑,E-mail:Lun.Yang@gmail.com;贺 林,E-mail:helin@bio-x.cn,Tel:(021)62933338
