儿茶素-腺嘌呤分子间相互作用的密度泛函研究
2014-03-19蔡皖飞李来才
蔡皖飞, 郑 妍, 李来才
(四川师范大学 化学与材料学院, 四川 成都 610066)
药物小分子与DNA之间的相互作用研究在化学、生物、医药等领域具有重要的意义.药物进入人体后一旦与DNA发生相互作用,就会影响到DNA的生理和物理化学性质,改变DNA的转录和复制[1].在医药研究中,DNA与药物分子相互作用的研究不仅可以阐述一些抗肿瘤、抗病毒药物及致癌物的作用机理,而且为新型药物的设计、修饰、合成和筛选提供有价值的信息,在针对DNA有显著识别功能的新型抗艾滋病和抗癌药物的开发中具有重要的意义[2-3].腺嘌呤(6-氨基嘌呤)既是构成DNA和RNA的碱基之一,也是人体中辅酶维B12的重要组成成分之一,腺嘌呤与磷酸结合物具有刺激白细胞的作用,广泛用于治疗各种白细胞减少症[4].同时腺嘌呤还用作探针分子,用于临床检测.
儿茶素是一种重要的含多个酚羟基的类黄酮化合物,现代医学研究表明[5-8]儿茶素具有抗氧化、抗癌、抗肿瘤和抑制DNA氧化损伤等功能,能够增强人体的免疫功能,抑制肿瘤的生长,对心血管疾病和艾滋病也有预防效果.研究人员通过光谱、电化学和理论计算等方法对其结构、抗氧化活性与结构和性能的关系等进行了研究[9-12].郭金保[1]采用荧光光谱法研究了儿茶素与DNA相互作用机制,发现当儿茶素与DNA相互作用时,振动频率向低波数方向发生了移动,即产生了红移.由于分子间的非共价相互作用较弱,实验上对于儿茶素与DNA相互作用的微观机制还未弄清楚.因此,采用量子化学方法对儿茶素与DNA碱基形成的复合物进行研究,对于了解这些活性小分子与生物分子的作用规律,进而阐明其生物学效应的机制无疑是有益的.我们研究小组在前期工作中对儿茶素与鸟嘌呤、胞嘧啶分子间相互作用进行了研究[13-14].本文通过对儿茶素-腺嘌呤复合物的研究,从分子水平上了解儿茶素与腺嘌呤分子间相互作用机制,为儿茶素类药物的设计、修饰、合成和筛选提供有价值的信息.
1 计算方法
采用密度泛函理论(DFT)B3LYP方法,在6-31+G*基组水平上对儿茶素、腺嘌呤及儿茶素-腺嘌呤复合物进行了几何优化,得到16种儿茶素-腺嘌呤复合物.并在相同基组水平上,应用分子中的原子(AIM)理论[15]和自然键轨道(NBO)理论[16]对儿茶素与腺嘌呤之间的相互作用特征和本质进行了分析.计算复合物相互作用能时,考虑了基组重叠误差(BSSE),采用S. F. Boys和F. Bernardi提出的完全均衡校(CP)在6-31+G*基组水平上进行BSSE校正[17].所有计算均采用Gaussian 03程序[18]完成.
2 结果与讨论
2.1儿茶素几何构型分析经过B3LYP/6-31+G*基组优化了5种同羟基取向的儿茶素构型,选取能量最低的构型作为研究对象.优化后的儿茶素几何构型如图1所示,儿茶素的几何优化参数及实验参数列于表1.由图1可知,儿茶素分子为非平面构型,在2个苯环上共有4个酚羟基团,可以参与氢键的形成,2个苯环通过一个含羟基的非芳香环连结.理论计算的构型参数和实验测定参数列于表1,表1数据表明计算值与实验值非常接近,键长平均误差小于5%,说明采用的计算方法是可信的.导致误差的原因可能在于理论计算的是气相单分子的结构参数,而本文中所采用的实验数据是在晶体状态下进行测定.
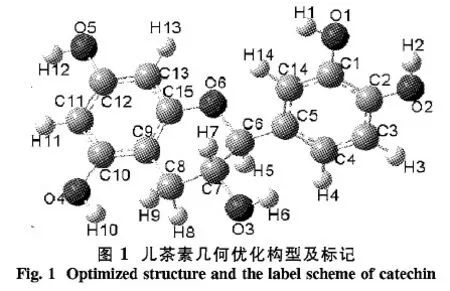
表 1 儿茶素的主要实验参数和B3LYP/6-31+G* 基组水平上的理论参数Table 1 Caculated and measured bondlengths of catechin and calculations were performed with B3LYP/6-31+G* nm

键型B3LYP/6-31+GExp.[19]键型B3LYP/6-31+GExp.[19]C1—O10.137 90.137 4C1—C20.140 60.140 1C2—O20.136 50.137 9C2—C30.139 20.142 1C7—O30.142 10.146 1C3—C40.139 90.138 5C10—O40.137 20.137 6C5—C60.151 10.152 8C12—O50.137 10.140 4C7—C80.152 60.155 5
2.2儿茶素-腺嘌呤复合物的几何构型分析采用B3LYP/6-31+G*基组对儿茶素-腺嘌呤复合物的构型进行全优化,得到稳定的儿茶素-腺嘌呤复合物16个.并在相同水平上进行了频率计算,所得构型均无虚频,证明其均是势能面上的极小点.优化得到的儿茶素-腺嘌呤复合物构型及结构参数如图2所示,相应的能量列于表2.研究结果表明儿茶素和腺嘌呤分子也会按照尽可能多的形成氢键相互作用这种几何优势来降低儿茶素-腺嘌呤复合物体系的能量,从而使其达到最稳定状态,其中儿茶素和腺嘌呤分子既是氢键供体又是氢键受体.氢键对儿茶素-腺嘌呤复合物的稳定性起着重要作用,同时我们还优化了π-π作用模式的儿茶素-腺嘌呤复合物,但没有得到稳定的构型.
为了弄清儿茶素-腺嘌呤复合物的成键情况,应用AIM2000程序计算复合物中键临界点的拓扑参数.根据Bader的AIM理论,P. L. A. Popelier等[20]提出氢键临界点处的电荷密度(ρ)在0.002 0~0.040 0 a.u.,相应的键临界点电荷密度的拉普拉斯值(▽2ρ)范围在0.024 0~0.139 0 a.u.,▽2ρ>0与闭壳层体系(离子键、氢键、Vander Waals)之间的相互作用有关,而▽2ρ<0则表征了在核间区域电荷密度是集中的共价键.从图2中可知,所研究的儿茶素-腺嘌呤复合物中只存在N—H…O、C—H…N和N—H…O等3种类型的氢键,所有形成的氢键X…H的临界点电荷密度均在0.008 0~0.040 0 a.u.之间,并且相应临界点处的电荷密度的拉普拉斯值在0.026 0~0.101 0 a.u.,符合氢键的定义.
作为一个普遍接受的观点,当分子间复合物只有一个氢键形成时,相互作用能的大小反映出氢键的强弱以及复合物的稳定性[21-22].但是,当复合物中存在2个或更多氢键时,对复合物稳定性及氢键强弱的判断将更加复杂[23].由图2可知,儿茶素-腺嘌呤复合物中除了复合物N形成了3个氢键外,其它的复合物都是形成了2个氢键.通过表2数据可知,复合物N是所有复合物中能量最低的,说明三氢键复合物N是最稳定的,氢键作用对儿茶素-腺嘌呤复合物稳定性起着重要作用.经BSSE校正后儿茶素-腺嘌呤复合物的相互作用能顺序为:N>B>P>O>A>C>J>E>G>M>K>D>L>F>H>I.由图2可知,最稳定的复合物N中存在N—H…O、C—H…N和O—H…N等3种类型的氢键,其键长分别为0.197 8、0.277 6、0.203 8 nm,对应的临界点电荷密度分别为0.027 8、0.008 0、0.020 5 a.u..形成的氢键使儿茶素和腺嘌呤分子稳定在处于近平面的位置,这表明儿茶素和腺嘌呤分子会按照尽可能多的形成氢键相互作用这种几


何优势来降低儿茶素-腺嘌呤复合物的能量,从而使其达到最稳定状态,所以形成三氢的复合物N相对其它复合物更具有几何优势.对于形成2个氢键的复合物,氢键的平均键长及电荷密度与复合物稳定性的顺序基本一致.只有复合物O和P除外,其平均键长分别为0.222 0、0.216 9 nm,对应的临界点电荷密度分别为0.016 8、0.018 4 a.u..而复合物A和C的平均键长分别为0.195 7、0.196 1 nm,对应的临界点电荷密度分别为0.028 5、0.027 5 a.u..可以看出复合物A和C的平均键长比复合物O和P的要小,对应的临界点电荷密度大,形成的氢键要强.可是复合物A和C的相互作用能却比复合物O和P的小,稳定性低,分析原因可能是其结构处于近平面的位置,类似与复合物N,虽然只形成了2个氢键,但是较其它复合物具有几何优势.对于形成2个氢键的这类复合物,复合物B是最稳定的,其形成的氢键N3…H2—O2、O1…H16—N2的键长分别为0.186 9、0.202 0 nm,对应的临界点电荷密度分别为0.035 5、0.022 1 a.u.,平均键长和电荷密度分别为0.194 4 nm和0.028 8 a.u,比其它此类复合物形成的氢键平均键长小,电荷密度大.表明复合物B的氢键作用更强,相互作用能更大.但是复合物B所形成的2个氢键并不是比这类复合物中其它复合物的2个氢键都强,这表明复合物的稳定性要综合考虑所有氢键的强度.另外,通过图1可知,除了复合物A、B、C、O、P,所有形成二氢键的复合物都含有O/N…H—C型氢键,由于O/N…H—C型氢键键长普遍较大(0.243 9~0.277 6 nm),电荷密度较小(0.008 0~0.011 6 a.u.),形成的氢键很弱,所以含有O/N…H—C型氢键的复合物稳定性较差,而不含O/N…H—C型氢键的复合物A、B、C、O、P的相互作用能明显比含这类氢键的复合物要大,稳定性要好.综合以上分析可知,当分子间相互作用时,复合物会尽可能多的形成氢键相互作用,这种具有优势的几何构型可以降低体系能量,增强体系的稳定性.当复合物中存在2个或更多氢键时,复合物的稳定性由所有形成的氢键类型以及强度共同决定.
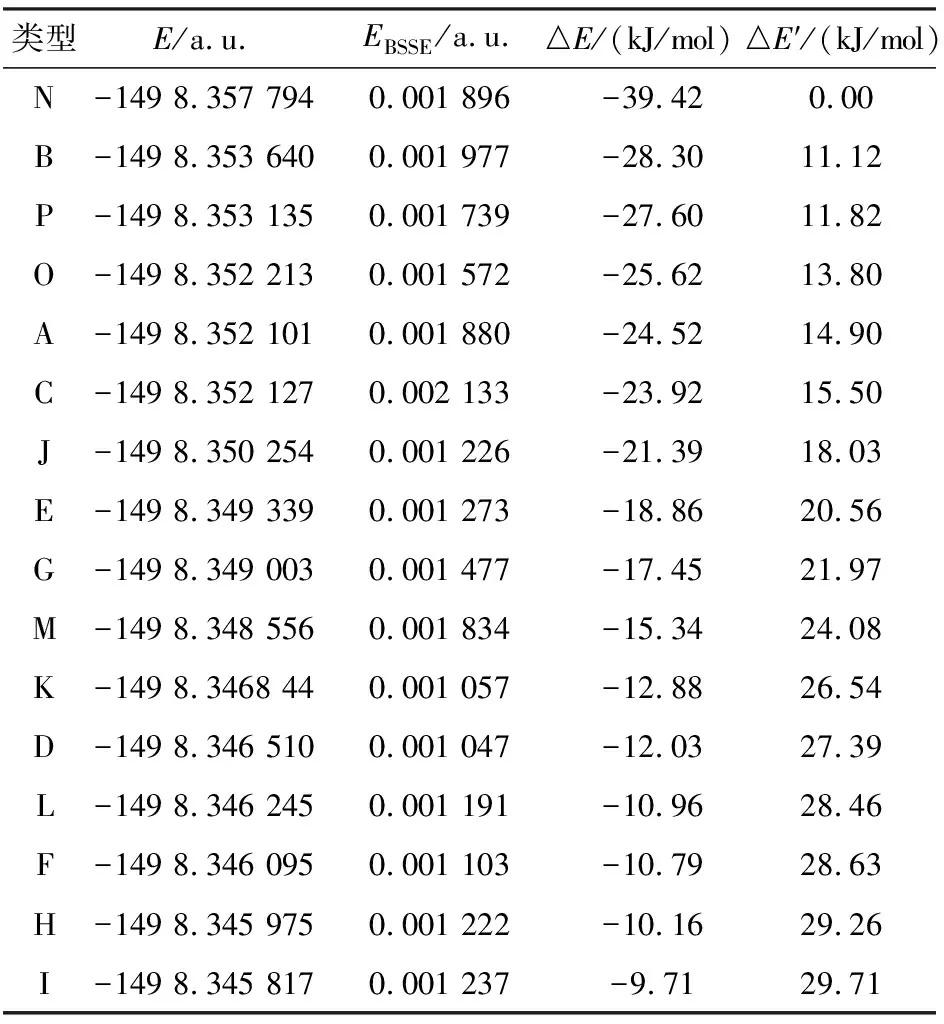
表 2 儿茶素-腺嘌呤复合物的总能量E、EBSSE、 EBSSE校正后相互作用能△E和相对能△E′Table 2 Calculated total energies E, EBSSE, interation energies with the EBSSE correction △E and the relative energies △E′ for all the complexes
2.3自然键轨道(NBO)分析为了揭示儿茶素-腺嘌呤复合物中氢键相互作用的本质,在相同基组水水平上对体系进行了自然键轨道(NBO)分析.表3给出了儿茶素-腺嘌呤复合物各构型中电子供体轨道i、电子受体轨道j及其相互作用的二阶稳定化能E(2),E(2)越大表明i与j的相互作用越强,即i提供电子给j的倾向越大,氢键作用越强.由表3及图2可知,儿茶素-腺嘌呤复合物中,腺嘌呤分子中N原子的孤对电子与其发生氢键作用的儿茶素分子中O—H反键轨道的相互作用较强,二阶稳定化能E(2)在47.80~94.40 kJ/mol范围内,形成的氢键作用很强;但是儿茶素酚羟基上氧原子上的孤对电子与其相互作用的腺嘌呤分子中N—H反键轨道的相互作用相对前者较弱,二阶稳定化能E(2)的范围为10.40~62.70 kJ/mol,这说明儿茶素和腺嘌呤分子既是氢键供体又是氢键受体,腺嘌呤分子中的N原子提供电子给儿茶素中O—H反键轨道的倾向要大,是稳定的电子供体.另外,通过分析发现腺嘌呤分子中N原子的孤对电子与儿茶素中相邻的C—H反键轨道的相互作用普遍较小,二阶稳定化能E(2)的范围为0.80~17.50 kJ/mol,形成的氢键较弱,表明儿茶素分子与DNA发生相互作用时,儿茶素分子中的羟基扮演着重要的角色.表3中数据表明同类型复合物的二阶稳定化能之和的顺序与相互作用能的顺序基本一致,但是复合物复合物O和P除外,分析原因与上述结构分析相同.对于形成三氢键的复合物N形成的3个氢键N3…H10—O4、N3…H10—C8、O3…H16—N2的二阶稳定化能分别为62.68、0.80、47.82 kJ/mol,二阶稳定化能之和为111.30 kJ/mol.对于形成二氢键的复合物,最稳定的复合物B的N3…H2—O2、O1…H16—N2的二阶稳定化能分别为90.83、40.58 kJ/mol,二阶稳定化能之和为130.41 kJ/mol,比其它的二氢键的复合物的要大.但是复合物B所形成2个氢键并不是比这类复合物中其它的复合物的每个氢键都强,这表明复合物的稳定性要综合考虑所有氢键的强度.同样对于形成二氢键的这类复合物,由于O/N…H—C型氢键二阶稳定化能普遍较小(0.80~17.50 kJ/mol),形成的氢键很弱,所以含有O/N…H—C型氢键的复合物稳定性较差,而不含O/N…H—C型氢键的复合物A、B、C、O、P的二阶稳定化能明显比含这类氢键的复合物要大,稳定性要好,这与前面的结构分析结果相一致.
2.4振动频率分析在B3LYP/6-31+G*水平计算儿茶素分子中O/C—H以及腺嘌呤分子中N—H伸缩振动频率,理论计算的儿茶素分子O—H伸缩振动频率值在3 716~3 770 cm-1,苯环上C—H伸缩振动频率值在3 187~3 233 cm-1,接近M. M. Ramos-Tejada等[11]的实验测定值3 600~3 200 cm-1和3 100~3 000 cm-1.误差产生的原因可能是因为儿茶素分子非平面的多环大分子,并且含有多个羟基,分子间易形成氢键作用;理论计算的为单个分子在气相中的参数,从而导致一定的误差存在,所以在结果对比中O—H伸缩振动频率的理论值与实验值相差要大,而苯环上C—H伸缩振动频率相差较小.从表4可知,当复合物形成氢键后,相应的O/C/N—H键均表现出伸长,变化范围在0.000 1~0.002 2 nm,振动频率明显减小,表现出正常的红移,这与实验结果相吻合[1].另外,表4数据表明N—H…O和O—H…N型氢键键长和频率变化比C—H…N型氢键要大.这可能是因为N和O原子上的孤对电子与相邻的O—H、N—H反键轨道相互作用强,这表明红移增大的程度与复合物相互作用能的变化趋势基本一致.
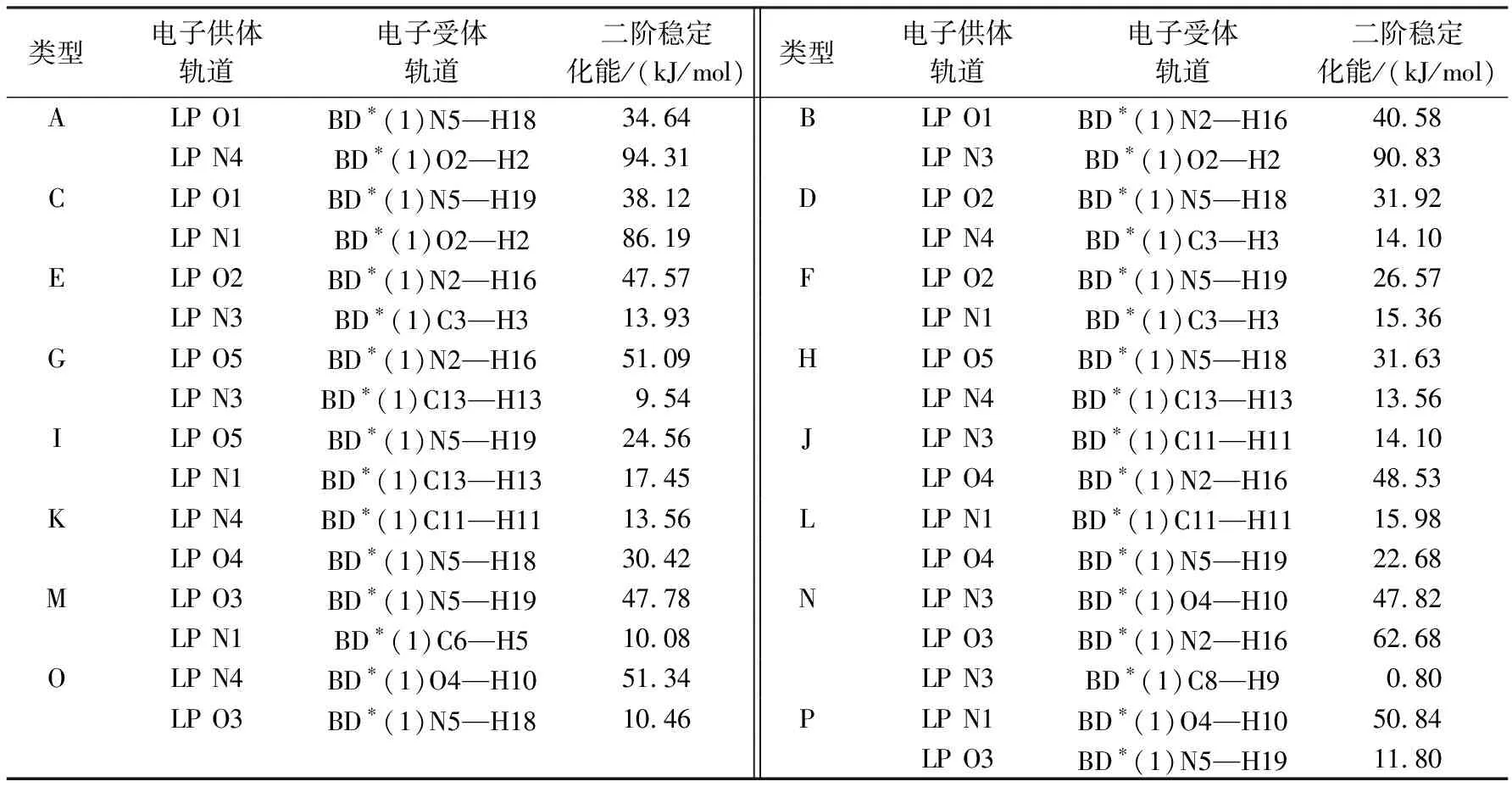
表 3 电子供体轨道和电子受体轨道以及相应的二阶稳定化能E(2)
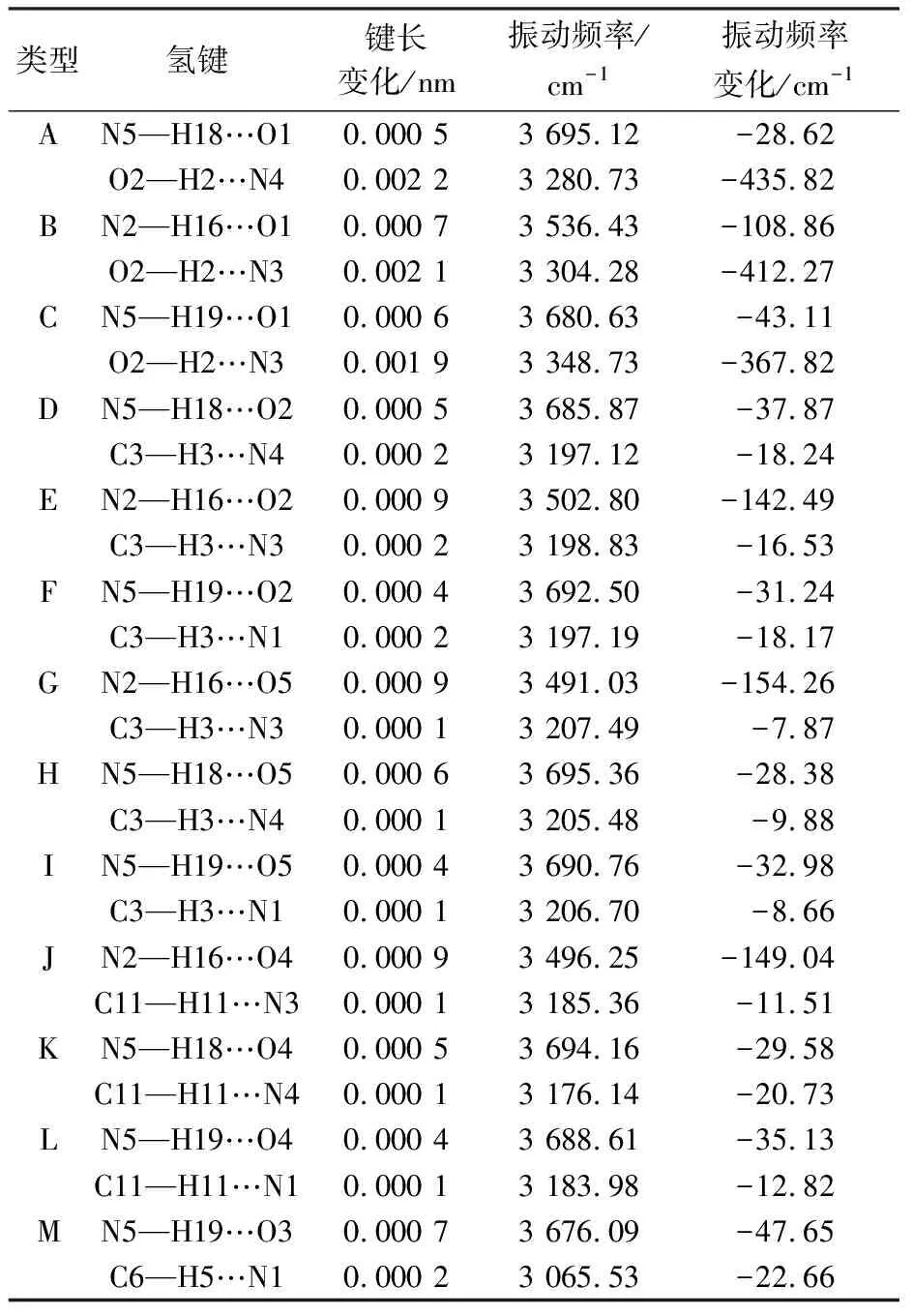
表 4 B3LYP/6-31+G*水平上形成氢键后X—H的 键长和相应的伸缩振动的变化Table 4 B3LYP/6-31+G* predicted changes in bond-length and stretching vibration of X—H bonds before and after hydrogen bond formation
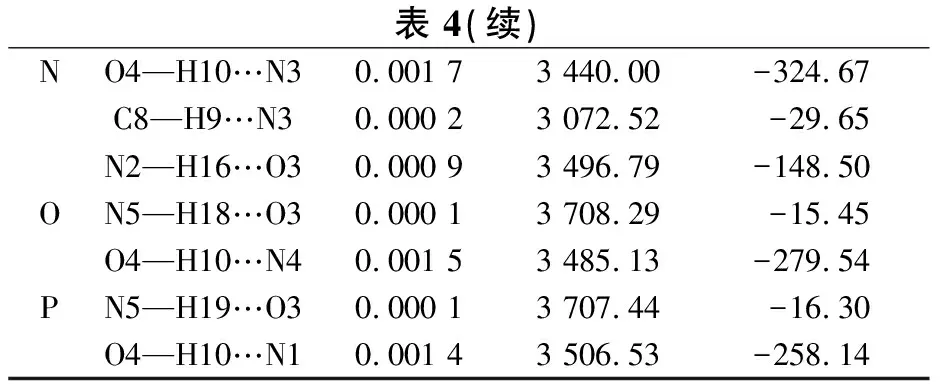
表 4(续)NO4—H10…N30.001 73 440.00-324.67C8—H9…N30.000 23 072.52-29.65N2—H16…O30.000 93 496.79-148.50ON5—H18…O30.000 13 708.29-15.45O4—H10…N40.001 53 485.13-279.54PN5—H19…O30.000 13 707.44-16.30O4—H10…N10.001 43 506.53-258.14
3 结语
采用密度泛函理论的B3LYP方法,在6-31+G*基组水平上研究了儿茶素与腺嘌呤的相互作用机制,得到稳定的儿茶素-腺嘌呤复合物16个,经BSSE校正后的相互作用能顺序为:N>B>P>O>A>C>J>E>G>M>K>D>L>F>H>I,优化后的构型和相互作用能分析表明儿茶素-腺嘌呤复合物中只存在N—H…O、C—H…N和O—H…N等3种类型的氢键,氢键对于儿茶素-腺嘌呤复合物的稳定性起着重要的作用.当儿茶素分子与腺嘌呤分子发生相互作用时,复合物会尽可能多的形成氢键相互作用来降低体系能量,增强体系的稳定性.通过振动光谱分析可知,儿茶素与腺嘌呤分子在形成氢键以后所有相应的O/C/N—H键均表现出伸长,振动频率明显减小,表现出正常的红移,且红移增大的程度与复合物相互作用能的变化趋势基本一致,这与实验结果相吻合.
[1] 郭金宝,张国文,陈秀霞,等. 儿茶素与DNA分子间的相互作用机制研究[J]. 分析科学学报,2008,24:507-511.
[2] 刘军,罗国安,王义明,等. 小分子与核酸相互作用的研究进展[J]. 药学学报,2001,36:74-78.
[3] Gorre M E, Mohammed M, Ellwood K, et al. Protein kinase inhibitors: insights into drug design from structure[J]. Science,2001,293:876-882.
[4] 陈新谦,金有豫. 新编药物学[M]. 12版. 北京:人民卫生出版社,1985:502.
[5] Yang B, Kotani A, Arai K, et al. Relationship of electrochemical oxidation of catechins on their antioxidant activity in microsomal lipid peroxidation[J]. Chem Pharm Bull,2001,49:747-751.
[6] Yang C S, Chung J Y, Yang G U, et al. Tea and tea polyphenols in cancer prevention[J]. J Nutr,2000,130:472-478
[7] Leanderson P, Faresjo A O, Tagesson C. Green tea polyphenols inhibit oxidant-induced DNA strand breakage in cultured lung cells[J]. J Free Radic Biol Med,1997,23:235-242.
[8] McKay D L, Blumberg J B. The role of tea in human health: an update[J]. J Nutr,2002,21:1-13.
[9] Teixeira S, Siqueta C, Alvesb C. Structure-property studies on the antioxidant activity[J]. J Free Radic Biol Med,2005,39:1099-1108
[10] Fronczek F R, Gannuch G, Mattice W L, et al. Dipole moment, solution, and solid state structure of (-)-epicatechin, a monomer unit of procyanidin polymers[J]. J Chem Soc PerkinTrans II,1984,2:1611-1616.
[11] Ramos-Tejada M M, Durn D G, Ontiveros-Ortega A. Colloids and Surfaces B:Biointerfaces,2002,24:297-308.
[12] 彭黎黎,刘靖丽,郭勇,等. 儿茶素结构和振动光谱的密度泛函理论研究[J]. 化学研究与应用,2008,8:1001-1006.
[13] Cai W F, Li L C, et al. Chin J Chem,2010,28(11):2137.
[14] 蔡皖飞,毛双,李来才,等. 儿茶素-胞嘧啶分子间相互作用的密度泛函研究[J]. 中国科学:化学,2011,41(6):1017.
[15] Bader R W F. Atoms in Molecules a Quantum Theory[M]. Oxford:Oxford University Press,1990.
[16] Reed A E, Weinhold F, Curtiss L A, et al. J Chem Phys,1986,84:5687-5695.
[17] Boys S F, Bernardi F. Some procedures with reduced errors[J]. J Mol Phys,1970,19:553-557.
[18] Frisch M J, Trucks G. W, et al. Gaussian 03, Revision A.7[M]. Pittsburgh PA:Gaussian, Inc,2003.
[19] Engle D W, Hattingh M, Hundt H K L. J Am Chem Soc Commun,1978, 6:95-102.
[20] Popelier P L A. Characterization of a dihydrogen bond on the basis of the electron density [J]. J Phys Chem,1998,A102:1873-1878.
[21] Kowalska A, Stobiecka A, Wysocki S. J Mol Struct:Theochem,2009,901:88-95.
[22] Mohajeri A, Nobandegani F F. Detection and evaluation of hydrogen bond strength in nucleic acid base pairs[J]. J Phys Chem,2008,A112:281-295.
[23] Lv G, Chen Z X, Zheng J, et al. J Mol Struct:Theochem,2009,939:44-53.
