一种快速构建集胞藻6803 petBD必需基因定点突变株的方法
2014-03-17芦亚菲曲娜陈晓波匡廷云
芦亚菲曲 娜陈晓波匡廷云
(1. 河北科技大学生物科学与工程学院, 石家庄 050018; 2. 中国科学院植物研究所, 光生物学重点实验室, 北京 100093)
一种快速构建集胞藻6803 petBD必需基因定点突变株的方法
芦亚菲1曲 娜1陈晓波1匡廷云2
(1. 河北科技大学生物科学与工程学院, 石家庄 050018; 2. 中国科学院植物研究所, 光生物学重点实验室, 北京 100093)
集胞藻6803(Synechocystis sp. Strain PCC 6803)是能够进行光合自养的原核生物, 因其遗传背景清楚, 特别是它具有天然的同源重组转化能力, 是研究光合作用的模式生物之一[1—3]。以集胞藻进行分子生物学研究, 最常用的方法是基因敲除, 即基于同源重组原理, 利用抗生素的抗性基因插入到目的基因内部, 使其功能丧失[4]; 如进行定点突变常规的方法是首先构建一个缺失质粒, 通过同源重组缺失目的基因的部分片段; 然后再构建一个回复突变质粒, 在回复质粒内部设计突变位点, 利用同源重组把缺失的基因片段(已经包含了突变的位点)再次重组进基因组, 与此同时引入第二种抗生素基因, 以第二种抗生素筛选转化子, 经连续筛选, 即可获得定点突变的转化子[5—7]。由于这种方法涉及基因的删除, 所以如果要对必需基因进行定点突变, 显然不能采用这种方法。
本研究在参阅相关研究文献[6—8]的基础上, 采用了一种改进的方法, 对集胞藻的必需基因pet BD基因进行了定点突变。Pet BD基因编码光合膜蛋白细胞色素 b6f蛋白复合体的细胞色素 b6和亚基Ⅳ这两个蛋白亚基, 两个基因之间有一段约200 bp的非编码区。该方法基本原理是在构建集胞藻同源重组转化质粒时, 在定点突变位点上游的非编码区内引入一个特异酶切位点, 经转化获得转化子后, 以引入的酶切位点作为第二个筛选标记鉴定转化子。在本实验中为获得 pet D基因的定点突变株,在其上游pet B和pet D之间的非编码区引入了一个HindⅢ酶切位点。本研究使用上述方法, 最终鉴定了三个 b6f蛋白复合体亚基Ⅳ的定点突变株, 单突Gly136Ala、单突Thr137Ala 以及双突Gly136Ala/ Thr137Ala。该方法是在集胞藻常规同源重组转化质粒构建的基础上稍加改进,克服了常规的基因敲除法不能构建必需基因定点突变株的缺陷, 并且可以快速预筛选出定点突变株, 为后续的测序节约成本。
1 材料与方法
1.1 实验材料与培养条件
集胞藻6803藻种为本实验室保存。集胞藻培养条件参照文献[1, 9], 突变株培养时在培养基中加入终浓度为25 μg/mL的卡那霉素。
宿主菌E. coli DH5ɑ用于质粒扩增的, 本实验室保存的质粒pET 30a, 宿主菌Trans-T1感受态细胞, pUC18质粒购自TransGen公司。
1.2 试剂与工具酶
ExTaq DNA Polymerase、dNTP Mixture、Pfu DNAPolymerase 购自TaKaRa公司; 限制性内切酶HindⅢ、EheⅠ、PstⅠ、EcoRⅠ购自Fermentas公司; DNA ligase、Trans Taq-T DNA Polymerase、Fast Mutagenesis System购自TransGen公司; 卡那霉素购自Sigma公司。DNA小提中量试剂盒, 琼脂糖凝胶回收试剂盒购自索莱宝生化科技(北京)有限公司。PCR引物合成以及DNA序列测序交由北京三博远志生物技术公司完成。
1.3 同源重组载体的构建
参照Putnam-Evans等[6,7]和Yan等[8]的方法构建载体,方法如下: 根据NCBI上提供的集胞藻6803petBD基因序列及其上下游序列和 pET 30a质粒中抗卡那霉素基因的表达框序列, 分别设计引物(Up-F、Up-R、Down-F、Down-R和 Kanr-F、Kanr-R; 表 1), 以集胞藻基因组和pET30a质粒为模板, 扩增出上、下游片段和抗性基因Kanr片段。按上游序列-Kanr片段-下游序列的顺序连接到pUC18质粒上。
采用 TransGen 公司的快速突变试剂盒(Fast Mutagenesis System), 选择在petB和petD基因之间的非编码区的一段序列AAGATT, 设计引物HindⅢ-F和HindⅢ-R(表 1), 进行一个碱基的突变, 引入 HindⅢ酶切位点(AAGCTT), 构建成pUC-petBD同源重组质粒。
根据集胞藻6803亚基IV的氨基酸编码序列设计三个氨基酸定点突变质粒的引物(G-F、G-R; T-F、T-R; D-F、D-R;表1), 采用TransGen公司的快速突变试剂盒, 以pUC-petBD质粒为模板, 构建三个氨基酸定点突变质粒, 即单突Gly136Ala、单突Thr137Ala 和双突Gly136Ala/Thr137Ala。
1.4 集胞藻6803的自然转化
集胞藻6803的自然转化按照Williams描述的方法[10]。
1.5 突变株的鉴定
以野生型集胞藻6803和三个定点突变株的基因组为模板, 分别用 K-F、K-R(表 2)引物鉴定 Kanr片段重组结果, HindIIIJD-F、HindIIIJD-R(表 2)引物鉴定引入的HindIII酶切位点是否已重组到集胞藻基因组中, 最后用petD-F、petD-R(表2)引物扩增petD片段, 进行测序验证。
2 结果
2.1 定点突变转化质粒的构建
同源重组质粒 pUC-petBD的构建图谱见图 1。为了验证构建的pUC-petBD载体上petB和petD之间是否引入HindIII酶切位点, 设计了引物HindIII JD-F和HindIII JD-R(表2), 用该引物对pUC-petBD载体进行扩增, 理论产物大小为616 bp, 酶切位点位于第216个碱基处。图2是对引入的酶切位点鉴定的电泳图, PCR产物大小为600 bp左右, HindⅢ酶切后产生大约200 bp和400 bp的2个片段, 与实验设计吻合, 说明成功地在petB和petD之间引入了一个新的HindⅢ酶切位点。

表1 构建同源重组载体定点突变载体所需的引物Tab. 1 Primer sequences for the construction of the site-directed mutagenesis plasmid based on homologous recombination
按照Transgen公司的Fast Mutagenesis System的操作, 以pUC-petBD质粒为模板, 根据集胞藻Cyt b6f亚基IV的编码序列分别设计引物(G-F、G-R; T-F、T-R; 表1),将petD的Gly136(GGC)突变成Ala(GCC)即单突Gly136Ala,Thr137(ACT)突变成Ala(GCT)即单突Thr137Ala。以及以单突Thr137Ala质粒为模板, D-F和D-R(表1)为引物构建双突Gly136Ala/Thr137Ala。这三个定点突变的质粒构建完成, 经测序验证正确(数据未显示)。

表2 PCR验证突变株所用的引物序列Tab. 2 Primer sequences for the identification by PCR

图1 同源重组载体pUC-petBD的构建图谱Fig. 1 Schematic describing the construction of the homologous recombinant vector pUC-pet BD

图2 HindⅢ酶切 pUC-petBD载体PCR产物的电泳分析Fig. 2 An electrophoretogram of HindⅢ-restricted from pUC-petBD vector
2.2 集胞藻6803的转化和Kanr突变株的筛选
将测序正确的三个定点突变质粒分别与野生型集胞藻6803细胞混合, 进行转化, 光照下孵育6h后, 涂布于不加卡那覆盖有硝酸纤维素薄膜的BG-11固体培养基上, 生长2—3d后, 将滤膜转到含有25 μg/mL 的卡那霉素固体板上培养。同时以野生型6803作对照。大约1周后, 野生对照没有长出转化子(图3a), 而三个重组平皿分别长出单菌落(图3 b, c, d)。
随机挑取转化子, 在25 μg/mL的卡那霉素BG11固体平板上进行抗性筛选, 通过 3—5次继代筛选后, 以野生型集胞藻 6803和三个定点突变株的基因组为模板, 用K-F和K-R为引物(表2)进行PCR扩增。结果表明在野生型集胞藻6803中的PCR产物约为0.3 kb,而在筛选的12个突变株的PCR产物都为约1.7 kb, 多出了约1.4 kb的抗性基因Kanr(图4)。这一结果表明, 在DNA水平上, 抗性基因已经重组到了集胞藻中。
2.3 定点突变株株的验证
以野生型集胞藻 6803和 12个成功重组抗性基因Kanr的突变株基因组为模板, HindⅢJD-F和HindⅢJD-R (表2)为引物进行PCR扩增。结果表明, 野生和12个突变株都能扩增出616 bp大小的DNA片段, 经HindⅢ酶切后, 野生型6803的PCR产物片段不能被切开, 12个突变株中有5株PCR产物能够切出约200 bp和约400 bp两条带, 其他 7株未被切开(图 5)。这一结果表明, 上述 5株(其 PCR产物能被 HindⅢ酶切开的突变株), 上游同源序列的重组至少开始于petB和petD之间的HindⅢ酶切位点, 在一般情况下, 其后的氨基酸突变位点也应该重组到集胞藻的基因组中。

图3 在含25 μg/mL卡那霉素的BG-11固体板上生长的转化子Fig. 3 Inducement of the transformants on BG-11 plate containing 25 μg/mL kanamycin (a) Wild type 6803; (b) Single mutant Gly136Ala; (c) Single mutant Thr137Ala; (d) Double mutant Gly136Ala/Thr137Ala

图4 鉴定突变株是否插入Kanr片段Fig. 4 Identification of the Kanrfragment insertion at DNA level
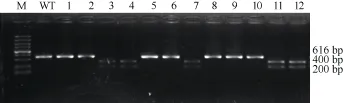
图5 鉴定突变株是否插入HindⅢ酶切位点Fig. 5 Identification of the Hind Ⅲ restriction site insertion at DNA level
以上述5株突变株(3、4、7、11、12)和另外7株(1、2、5、6、8、9、10)为模板, 用测序引物petD-F和petD-R(表2), 扩增 petD基因, 对 PCR产物进行测序验证, 结果表明上述5个突变株确实发生了定点突变(测序结果未显示),其中3、4为单突G136A, 7为单突T137A, 11、12为双突G136A/T137A。而在其他7株当中, 经测序验证只有1株发生了定点突变, 另外 6株并未发生定点突变(测序结果未显示)。
3 讨论
集胞藻是光合作用等研究领域常用的一种模式生物, 它具有天然的同源重组特性[1—3]。采用分子生物学方法对目的蛋白进行功能研究时, 对于非必需基因, 无论是进行基因敲除还是定点突变, 已经有了大量的研究,方法已经非常成熟[4—7]。但是对于编码细胞色素 b6f 的亚基Ⅳ蛋白的petD必需基因进行定点突变, 相关的研究报道不多。
在同源重组载体上游区域或下游区域设计突变位点,直接转化集胞藻, 因为重组的起始和终止位点难于确定,无法保证突变位点能够与基因组发生重组, 如果盲目地对每个转化子进行测序, 无疑会加大工作量和成本。但是如果在突变位点上游或下游(如果是突变位点位于转化质粒下游区域的情况)的非编码区提前设计一个新的酶切位点, 本实验中在Cyt b6亚基(petB编码)和亚基Ⅳ(petD编码)之间的非编码区对一个碱基进行突变, 形成一个新的HindⅢ的特异酶切位点。用该质粒转化得到转化子后, 扩增该酶切位点上下游区域, PCR产物经酶切可以鉴定该酶切位点是否重组进转化子, 因为该 HindⅢ酶切位点在petD基因的上游, 显然如果这一位点重组进了集胞藻基因组, 那么其下游的突变位点也应该重组到野生 6803基因组中。因此在突变位点之前非编码区引入特异酶切位点,可以看作是第二个筛选标记。
在本实验中, 重组在引入的酶切位点之前发生的概率是比较高的, 实验挑选了12个转化子, 其中 6个发生了定点突变, 其中有 5个鉴定为重组发生在引入的酶切位点之前, 这可能是与引入的酶切位点与突变位点距离较近和该酶切位点上游的同源序列较长有关。上游同源序列总长约1.7 kb, 引入的HindⅢ酶切位点约在1.0 kb的位置, 定点突变的位点约在1.4 kb的位置, 则酶切位点之前的同源区为约1.0 kb, 其与定点突变的位点相差约0.4 kb。即使这样, 在实验中仍发现了虽然抗性基因 Kanr已经重组到基因组中, 但是 HindⅢ酶切反应成阴性, 并且测序也发现未发生定点突变的情况(挑选的12个转化子中有6个属于这种情况)。不难想象, 如果遇到突变位点(或者是引入的酶切位点)之前的同源序列较短的情况, 这种方法的优势可能会更加显著。因为在这种情况下, 同源重组发生在突变位点下游的概率就会增加, 即突变位点的序列不发生同源重组, 那么利用突变位点之前引入的酶切位点为筛选标记, 然后对酶切反应呈阳性的转化子进行测序, 这样就避免了后续实验的盲目性, 大大提高筛选的效率。
综上所述, 应用本方法对petD这样的必需基因进行定点突变, 只需构建一次质粒, 进行一次转化, 剩下的工作主要是应用常规的PCR和酶切对抗生素平板上的转化子进行验证, 实验方法简单。该方法不但克服了常规方法不能用来构建必需基因定点突变的缺点, 并且可以简单快速预筛选出定点突变株, 对其他必需基因定点突变的研究具有借鉴意义。
[1] Yan C L, Xu X D. Isolation and characterization of mutants impaired in photoautotrophic growth in Synechocystis sp. PCC6803 [J]. Acta Hydrobiologica Sinica, 2009, 33(2): 360—362 [阎春兰, 徐旭东. 集胞藻 6803光合自养生长突变株的筛选与鉴定. 水生生物学报, 2009, 33(2): 360—362]
[2] Fu J, Xu X D. A priA mutant of Synechocystis sp. PCC6803 [J]. Acta Hydrobiologica Sinica, 2008, 32(1): 116—119 [付娟, 徐旭东. 集胞藻PCC6803priA 基因的突变体. 水生生物学报, 2008, 32(1): 116—119]
[3] Long Z J, Wang Q X, Ma W M, et al. Construction of the ndhO gene inactivation mutant of the cyanobacterium Synechocystis sp. PCC6803 [J]. China Biotechnology, 2010, 30(9): 31—35 [龙宗娟, 王全喜, 马为民, 等. 利用同源重组构建蓝藻集胞藻 6803ndhO基因突变株及其分子鉴定.中国生物工程杂志, 2010, 30(9): 31—35]
[4] Gao H, Tang Q, Xu X D. Construction of copper-induced gene expression platform in Synechocystis sp. PCC6803 [J]. Acta Hydrobiologica Sinica, 2007, 31(2): 240—244 [高宏,唐蜻, 徐旭东. 集胞藻 PCC6803铜离子诱导表达平台的构建. 水生生物学报, 2007, 31(2): 240—244]
[5] Wang M, Shan J X, Kuang T Y, et al. Advances in the research of structure and function of photosystemⅡ core antenna complexes CP43 and CP47 [J]. Chinese Bulletin of Botany, 2000, 17(2): 141—149 [王梅, 单际修, 匡廷云, 等.光系统Ⅱ核心天线复合物CP43和CP47结构与功能研究进展. 植物学通报, 2000, 17(2): 141—149]
[6] Cindy Putnam-Evans, Jituo Wu, Terry M Bricker. Site-directed mutagenesis of the CP 47 protein of photosystem Ⅱ: alteration of conserved charged residues which lie within lethal deletions of the large extrinsic loop E [J]. Plant Molecular Biology, 1996, 32(6): 1191—1195
[7] Cindy Putnam-Evans, Terry M Bricker. Site-directed mutagenesis of the CPa-1 protein of photosystem II: alteration of the basic residue pair384,385R to384,385G leads to a defect associated with the oxygen-evolving complex [J]. Biochemistry, 1992, 31: 11482—11488
[8] Yan J, Dashdorj N, Cramer W A, et al. On the structural role of the aromatic residue environment of the chlorophyll a in the cytochrome b6f complex [J]. Biochemistry, 2008, 47(12): 3654—3661
[9] Allen M M. Simple conditions for growth of unicellular blue-green algae on plates [J]. Journal of Phycology, 1968, 4(1): 1—4
[10] Williams J G K. Construction of specific mutations in photosystem Ⅱ photosynthetic reaction center by genetic engineering method in Synechocystis 6803 [J]. Methods in Enzymology, 1988, 167: 766—778
A RAPID METHOD FOR SITE-DIRECTED MUTAGENESIS OF THE ESSENTIAL GENE PETBD IN SYNECHOCYSTIS SP. PCC 6803
LU Ya-Fei1, QU Na1, CHEN Xiao-Bo1and KUANG Ting-Yun2
(1. College of Biological Science and Engineering, Hebei University of Science and Technology, Shijiazhuang 050018, China; 2. Key Laboratory of Photobiology, Institute of Botany, Chinese Academy of Sciences, Beijing 100093, China)
定点突变; pet BD必需基因; 酶切位点; 集胞藻6803
Site-directed mutagenesis; Pet BD essential gene; Restriction site; Synechocystis sp. PCC 6803
Q784; Q-33
A
1000-3207(2014)05-0957-05
10.7541/2014.141
2013-07-01;
2014-02-12
国家“973”项目(2011CBA00901)资助
芦亚菲(1988—), 女, 河北石家庄人; 硕士生; 细胞色素b6f复合物的结构与功能研究。E-mail: 630540631@qq.com
陈晓波(1975—), 男, 河北邯郸人; 博士; 主要从事细胞色素b6f复合物的结构与功能研究。E-mail: zzschenxiaobo@163.com
