气候变化背景下极地海洋和陆地生态系统中持久性有机污染物的迁移和分布
2014-03-08武晓果谢周清
武晓果 谢周清
(1安徽师范大学环境科学与工程学院,安徽芜湖241000;2中国科学技术大学极地环境研究室,安徽合肥230026)
0 引言
持久性有机污染物(persistent organic pollutants,POPs)是一类有毒的、在环境中难以降解且容易随着食物链富集以及常常随大气、水体和迁移物种长距离传输的化学物质[1-2]。POPs一旦被排放进入环境,便会在不同的环境介质中广泛分布,比如土壤、水、大气、大气气溶胶、植被和冰雪[3-5]。由于POPs物质具有持久性、半挥发性的特点,大气往往是其传输的主要途径,这也导致POPs很快随着大气传输而成为一种全球性的污染,而不仅仅是限于其历史使用地区[5-6]。POPs的地球化学循环首先受到其物理化学性质的影响,包括挥发性、水溶性、在环境介质中的半衰期等。另一方面,POPs的地球化学循环还受到环境因素的影响,例如温度、降水情况以及大气环流情况等。考虑到POPs物质具有半挥发性的特点,在诸多环境因素中,全球气候变化导致的温度波动是影响POPs地球化学循环的关键。这其中包括温度变化的直接影响,例如导致POPs直接从土壤等环境介质中的挥发[7];以及温度变化导致的间接影响,例如冰川(山川或者极地)覆盖面积的减少而导致的POPs界面交换作用的变化[4]。
POPs历史使用往往集中在工农业发达的北半球温/热带地区,这一地区POPs逃逸进入大气的趋势较明显。之后伴随大气传输,POPs倾向于在温度较低的高纬度地区沉降,极地成为一个“冷肼”而富集大量POPs物质[8]。一旦极地温度发生变化,随之而来的陆地冰川消退、海冰消融等环境事件会使得这些被捕集的POPs物质再次挥发进入大气中,之前的POPs环境储库转变成为POPs的二次来源[9]。另外一方面,低纬度源区的土壤是POPs重要的环境储库,在气温升高的背景下,这些富集在土壤中的POPs会更加容易再次恢复进入大气,通过传输到达极地,极地POPs的来源增加。同时,温度的升高也会导致半挥发性的POPs在大气中的分配状态发生变化,在大气中更容易以气态,而不是以颗粒态存在,使得POPs的传输距离增加,也更加容易到达极地[9]。在POPs被广泛限制和禁止使用的背景下,可以预料一次污染源(直接排放的POPs物质传输到达极地)正明显的减少,而这种由温度波动引起的二次排放会对南北极地区脆弱的生态系统造成较大风险。
1 物理化学性质对极地POPs迁移、分布的影响
POPs的地球化学循环与其物理化学性质有直接的关系[10-11],其中关键性参数有:(1)蒸气压(po,Pa)或者是辛烷-大气分配系数(KOA),常常用来指示POPs物质的挥发能力;(2)亨利常数(HLC,Pa·m3·mol-1)或者是大气-水分配系数(KAW),常常用来指示POPs物质在大气和水体之间的分配情况;(3)水溶性(g·m-3)或者是辛烷-水分配系数,常常用来描述POPs污染物质在水生生态系统中的分配情况[11-14]。
不同的理化性质决定了POPs的传输方式不同,一般而言对于极地POPs主要的传输方式有:(1)大气传输;(2)洋流传输;(3)随着生物迁徙而传输(图1,2)。当然在特殊的情况下POPs也可能随着漂移的海冰,或者大型河流的输入而进入极地。但是如前所述,鉴于POPs半挥发性,以及疏水性的特点,大气传输是占据主导的长传输方式[10]。这就说明KOA较低或者po较高的POPs会迅速从中低纬度的土壤中挥发,随着大气循环到达极地,同时这些POPs也更加容易从极地的土壤中逃逸进入大气。HLC或者KAW较高的POPs会更加容易从中低纬度水体中逃逸,随大气进入极地,但也更容易从极地水体逃逸,这对于海洋面积较大的北极而言是不容忽视的(图1)。这似乎说明若是极地温度发生波动,响应最为明显的应该是KOA较低或者HLC较大的POPs。例如HLC较大的六氯苯(hexachlorobenzene,HCB)和多氯联苯(polychlorinated biphenyls,PCBs),以及KOA较低、挥发性较强的α-六六六(α-hexachlorocyclohexane,α-HCH)(表1列出了一些常见POPs的KOA以及HLC)。在真实环境中,POPs的地球化学循环过程往往不能只依靠其物理化学参数来解释,其他的因素,例如(1)各种POPs物质历史使用情况、残留情况以及源区分布情况;(2)POPs在传播过程中的降解程度;(3)POPs在不同环境介质中的驻留程度,以及在不同环境介质中的交换情况;(4)环境因素,诸如温度、植被覆盖等的波动,将共同影响其环境命运。下文中分别探讨气候变化背景下极地海洋生态系统和陆地生态系统中POPs的分布和迁移。
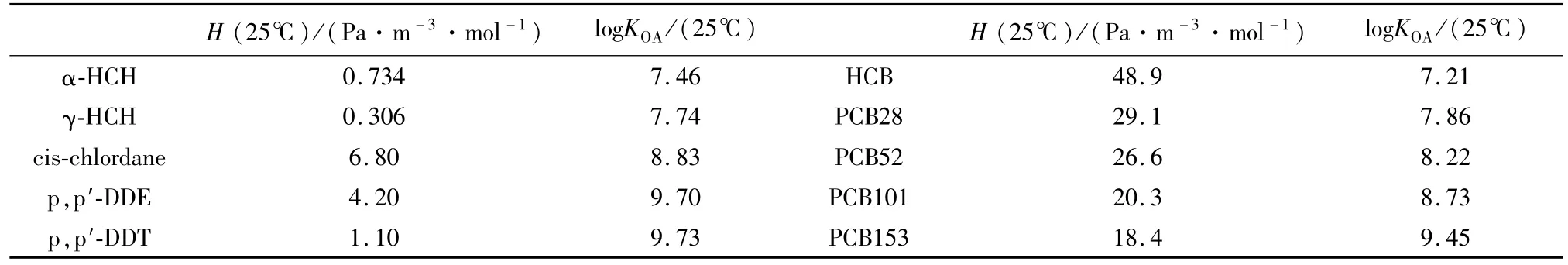
表1 典型POPs的物理化学性质[15]Table 1.Physical-chemical properties of typical POPs[15]
2 气候变化背景下极地海洋生态系统中POPs的迁移、分布
海洋是全球污染物最大的储库,POPs也不例外,研究者已经做了很多工作,在海水、海冰以及海洋大气中均检出了 POPs的存在[4,16-31]。考虑到一些POPs物质已经被禁止或者限制使用超过30年以上[1-2],从使用地区经过长距离传输而对极地的贡献可能会逐渐减弱。由于POPs的环境持久性,以及极地的冷肼效应,很长一段时间内它们可能会在极地内部循环,尤其是在不同环境介质之间。对极地海洋生态系统而言,其地球化学循环中有如下三个环节是至关重要的:(1)表层海水和底层大气之间的交换;(2)海冰和底层大气之间的交换;(3)表层海水和深层海水之间的交换。
2.1 气候变化背景下POPs在极地表层海水-底层大气之间的交换
针对表层海水-底层大气之间的交换,目前业已开展了相当多的研究,但是值得注意的是主要研究区域集中于北极,南极海域相关的研究较为缺乏。针对不同的POPs,它们的逃逸趋势有所不同,如上节所述,这和不同POPs物理化学性质的差异相关,同时也和环境因素,在极地地区的残留总量相关。目前相关研究中发现逃逸趋势较为明显的POPs,往往是历史上使用量较大的POPs,例如α-HCH。历史上 α-HCH的排放量大约是 5 800—6 800 kt[32-33]。从20世纪70年代开始逐步被淘汰以后,北极大气中α-HCH的浓度表现出降低的趋势(北半球为HCH主要使用地区)[16]。随之北极地区α-HCH交换的状态也发生了变化。在使用期,源区的α-HCH不断通过大气长距离传输到达较冷的北极,表层海水-底层大气之间的交换方向以从大气进入海洋为主导,即海洋为α-HCH的汇[5-6]。从20世纪90年代初至21世纪,很多在北极的研究均发现α-HCH有明显从表层海水向海洋大气逃逸的趋势,中国2003年和2008年北极考察的观测也发现了类似的结果,说明北极内部传统的汇正转变成α-HCH的源[16,21-22,34-37]。不单单是 α-HCH,随着 POPs的限制,可以预见二次来源的POPs对极地生态系统的暴露将会越来越占据主导,尤其是一些KOA较低或者HLC较大的POPs,例如上文中提到的PCBs以及HCB,以及历史使用量较大的其他POPs,例如DDT及其降解产物。
而对于北极广阔的海域来说,海气交换作用是重要的环节。首先,POPs的HLC受到温度的影响,随着温度的增加HLC有增大趋势[38]。这说明气温的升高,导致HLC的变化驱使着海水中的POPs有更大的向大气中逃逸的趋势。但是另一方面,温度升高使得高纬度地区降雨量增大,导致极地海洋盐分的减少,这会导致海水对POPs的溶解度增大(盐析),从而造成POPs向海水中沉降的趋势增加[9]。具体以哪一种变化方向为主受到多种环境因素的影响,也受到不同POPs种类的影响,目前还没有具体的研究提出其具体变化量。研究者利用大气-界面扰动耦合模型对北极环境中大气-海面之间的交换情况进行了拟合,结果表明在气候变化的背景下,北极大气中的α-HCH、PCB28、PCB52以及 PCB101浓度水平将会持续增长,并且在较高水平一直到大约2037年,之后随着在环境介质中的降解作用,大气中浓度水平才会回落[15]。这样的模型预测结果和北极地区长期监测的结果也是吻合的[39]。说明海水中的逃逸也是造成POPs活跃的一个重要原因。但是针对水溶性较高的POPs,例如γ-HCH和PCB153,它们的释放作用会持续很久的一段时间,这和他们在环境储库中的稳定性和贮存时间相关[15]。
其次,极地的海气交换趋势无疑受到海冰覆盖的巨大影响。海冰的存在使得表层海水-底层大气之间的交换被停止,在海水凝结海冰时,POPs往往容易留存在海水中,这使得海冰下海水富集了大量的POP,有研究表明一般海冰下水中POPs逃逸趋势要比平衡状态高2—5倍[40],这说明一旦海冰消退,海气的交换作用会再次活跃,且往往以从海水中的释放为主要方向。这种现象更直接的证据来自夏季海冰边缘地区的监测,例如2008年中国北极考察[16]以及 Gioia等人[20]报道的 2004年北极考察的结果,分别发现α-HCH和PCBs在海冰边缘区域明显高于海冰覆盖区。此外,还有另外一种情况可能导致海冰边缘区域POPs浓度的增高,即POPs物质直接从消融的海冰中进入大气,这将在下节中论述。
2.2 气候变化背景下POPs在极地海冰-大气之间的交换,以及海冰消融对极地POPs海-气交换的影响
极地冰雪样品中POPs的检测工作开展得较少,但是仍然获得了一些结果,说明大气沉降会导致海冰(积雪)中富集 POPs[4,19,27],这其中包括气态POPs在冰雪表面的富集,以及降雪冲刷下来的大气中附着在颗粒(气溶胶)上的POPs[4]。作为临时储库的季节性冰川/海冰消退的时候,会导致整个冬季储存在冰雪中的POPs物质在很短的夏季消融时段释放出来,使得极地大气中POPs浓度水平临时突然陡增。而这种增加的趋势和极地冬季降雪量的多少,积雪时段的长短,以及夏季积雪/冰川消融量的多少密切相关。另一方面,随着气候变化,一些多年生海冰也出现了逐年减少的趋势,从20世纪70年代起,北极海冰覆盖面积每十年呈现2.3%的下降。而这些多年生海冰由于形成时期较早,可能富集POPs量较季节性海冰要高。此外,较之季节性海冰,多年生海冰较为密实,空隙较少。一般而言,极地降雪的比表面积大约为0.1m2·g-1,伴随着冻融的过程,其表面积急剧减少至0.001 m2·g-1以下,最终形成密实的多年生海冰[40]。较小的比表面积意味着较高的逃逸趋势,一旦多年生海冰消融,其中的POPs会比季节性海冰更加迅速地逃逸进入大气,或者是在融水中POPs的逃逸趋势较大,这些都会造成极地大气POPs水平的迅速升高。因此,海冰的消融也是极地大气POPs不可忽视的二次来源。
如上节所述,海冰的年季变化和季节性波动还会对极地POPs表层海水-底层大气交换造成影响。对比中国2003、2008和2010年北极考察的结果可以发现,大气中α-HCH的浓度在2008年最高,且α-HCH的界面交换趋势在2010年以底层大气向表层海水沉降为主[16,37,41]。这种现象似乎和 20世纪90年代以后很多研究发现的α-HCH以从表层海水逃逸进入底层大气为主的现象相反[16,37,41]。但是结合北极海冰的变化趋势来看,之前中国北极考察的研究结果是可以解释的。根据美国冰雪数据中心的海冰监测数据(http://nsidc.org),2008年海冰覆盖面积是以上三个年份中最少的,说明2008年考察期间α-HCH无论是从海冰中的释放还是从海水中的释放均会较强,因此大气中α-HCH的浓度在2008年最高。另一方面,2008—2010年间北极的海冰有明显增加的趋势,因此和2008年比较,2010年北极考察时α-HCH从北极海冰和海水中的释放作用较弱,甚至会出现逃逸趋势相反的现象。综合来说,不同年份的冰情分布会使得从积雪/冰川中逃逸出的POPs量出现差异,同时也会使得POPs海气交换方向以及交换通量出现差异,这些作为气候变化的结果,共同影响着POPs在极地的分布和循环。
2.3 气候变化背景下极地海洋(海水)中POPs的迁移、分布
对于海洋中POPs的地球化学循环来说,生物泵作用(biological pump)是关键的环节。海洋浮游植物通过光合作用吸收大气二氧化碳、释放出氧气,成为海洋食物链中其他各级生物的有机质食物来源。而大量由生物形成的含碳微粒,如粪便和微生物尸体等从海洋的表层沉降到深海。而POPs由于具有疏水性的特征,在海水中往往倾向于附着在这些含碳微粒上而沉降于深海中[42]。这个过程使得大气中的POPs不断通过生物泵作用进入深海,从而降低了POPs对海洋生态系统的暴露风险。可以预见到,通过生物泵作用,表层海水中POPs浓度降低,这也驱使着大气中的POPs向海洋沉降,从而进入深海[42]。
全球气候变化背景下,极地海洋生态系统遭受巨大的冲击,从而也影响着POPs通过生物泵作用在极地海洋中的沉降。首先,海洋初级生产力会受到气候变化导致的海水温度增加的影响。研究表明,自20世纪80年代早期以来,全球海洋的年初级生产力至少减小了6%,其中有将近70%发生在高纬度的南北极地区[42-43],这意味着气候变化导致极地海洋中浮游植物生物量减少。另一方面,海冰对维持极地海洋的生物多样性具有重要作用。春季融化决定着水华爆发的时间,对极地海洋食物网的动态也具有影响作用。极地海洋的食物网是以海冰藻类为基础的,所以海冰的消失也会使得海冰藻类生物量减少,进而导致极地海洋初级消费者和次级消费者生物量减少。例如研究发现,海冰消失可能是导致极地磷虾剧烈减少的主要原因(10年减少75%±21%)[43]。从以上两点来看,全球气候变化导致的极地海洋浮游植物生物量减少和海冰消失导致的海冰藻类减少无疑会导致极地海洋中含碳微粒减少,使得正常的生物泵作用被消弱,进而导致大量的POPs物质难以通过生物泵作用进入极地深海,而是在表层海水以及极地大气之间不断循环。这会显著增加极地生物被POPs暴露的风险。
3 气候变化背景下极地陆地生态系统中POPs的迁移、分布
对于温度较低的极地而言,POPs的循环更加类似于一种内部循环,这点是在之前的很多研究中都有发现的。虽然极地温度变化的总体区间较为狭窄,但是这种温度变化导致的POPs循环的变化却也不容忽视。在南极Livingston岛的研究结果表明,南极大陆温度变化区间只有大概4℃左右,但是PCBs的浓度仍然和温度之间有密切的联系,随着温度的升高,大气中PCBs的浓度表现出增加的现象[44]。
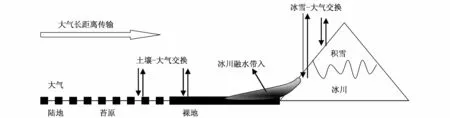
图2 极地陆地生态系统POPs迁移和分布示意图Fig.2.Distribution and transportation of POPs in the Polar Terrestrial Ecosystems
在极地陆地生态系统中,大量POPs被富集在土壤(包括纬度较低的极地苔原以及纬度较高的裸地)和极地冰川中。与海洋生态系统中海冰的作用类似,气候变化导致的极地陆地冰川的消融会导致其中富集的POPs物质再次释放进入大气中,增加对极地生态系统的暴露风险,这点不再赘述。模型的研究表明,在全球气候变化的背景下,0.05—0.1 K的温度增加,就可能导致土壤中的POPs物质加速向大气释放,而使得大气中POPs浓度增加4%—50%[45]。同时,冰川融水进入极地动物聚居地,也会增加对其的暴露。这种现象已经在前人的研究中有所察觉。如在南极区域收集的阿德利企鹅(Adélie penguin)样品的研究中,发现企鹅皮下脂肪组织中DDTs浓度在2004—2006年有升高的趋势。由于企鹅生活区域仅仅限制在南极地区,不会有接触到新鲜源的机会,因此可以判断是受到北极地区DDTs暴露的影响,其来源推测便是由于冰川融水使得之前沉降的DDTs有了再次产生暴露的可能[46]。
Livingston岛的研究还发现PCBs的逃逸还与另外一个因素密切相关,这就是土壤的有机质含量。温度的增加导致极地土壤中POPs逃逸趋势的增加,但是土壤有机质的增加却抑制了这种逃逸趋势。而这其中关键的一点是温度的升高会导致苔原面积扩大,苔原向高纬度发展,导致之前为裸地的土壤中有机质含量增加[44]。此时POPs的地球化学循环无疑是受到温度增加,以及其导致的土壤有机质含量增加共同作用的影响。而究竟这种耦合作用的贡献方向是如何的,以及定量的交换通量如何,还都是需要继续研究的问题,这可能是掌握在全球温度变化背景下POPs在极地大陆生态系统循环的关键。
4 结论
近十年来极地POPs表现出活跃的趋势,多方面的研究表明气候变化以及其导致的极地环境因素的变化,是导致POPs活跃的重要原因。本文分析了气候变化背景下,极地海洋生态系统和陆地生态系统中POPs的迁移和转化。需要看到的是极地POPs界面交换趋势和交换通量的定量研究,以及气候变化背景下与温度相关的一系列耦合作用对POPs地球化学循环的影响仍然是以后研究的重点。这些问题的解答对深入理解极地POPs地球化学循环,从而对其生态风险做出正确评估具有重要意义。
1 Stockholm Convention.Global Monitoring Plan for Persistent Organic Pollitants,First Regional Monitoring Report:Asia-Pacific Region.Stockholm Convention,2008.http://chm.pops.int/Portals/0/Repository/GMP/UNEP-POPS-GMP-RMR-ASIAPACIFIC.English.PDF.
2 Stockholm Convention.Global Monitoring Plan for PersistentOrganic Pollutants,First RegionalMonitoring Report:Western Europe and Other States Group(WEOG)Region.Stockholm Convention,2009.http://chm.pops.int/Portals/0/Repository/GMP/UNEP-POPS-GMP-RMR-WEOG.English.PDF.
3 Simonich S L,Hites R A.Global distribution of persistent organochlorine compounds.Science,1995,269(5232):1851—1854.
4 Wania F,Halsall C.Air/snow exchange of persistentorganic pollutants.Canadian Arctic Contaminants Aeeseement ReportⅡ,Volume 5:Sources,Occurrences,Trends and Pathways in the Physical Environment;Northern Contaminants Program:Hull,Quebec.2003:185—190.http://caid.ca/Vol5_Arct_Cont_2003.html.
5 Wania F,Mackay D.Tracking the distribution of persistentorganic pollutants.Environmental Science&Technology,1996,30(9):A390—A396.
6 Scheringer M.Long-range transport of organic chemicals in the environment.Environmental Toxicology and Chemistry,2009,28(4):677—690.
7 Meijer SN,Shoeib M,Jones K C,etal.Air-soil exchange of organochlorine pesticides in agricultural soils.2.Laboratorymeasurementsof the soilair partition coefficient.Environmental Science&Technology,2003,37(7):1300—1305.
8 Mackay D,Wania F.Transport of contaminants to the Arctic:Partitioning,processes and models.Science of the Total Environment,1995,160-161:25—38.
9 Lamon L,Valle M D,Critto A,etal.Introducing an integrated climate change perspective in POPsmodelling,monitoring and regulation.Environmental Pollution,2009,157(7):1971—1980.
10 AMAP.AMAPAssessment2002:PersistentOrganic Pollutants in the Arctic.Arctic Monitoring and Assessment Programme(AMAP),2004,Oslo,Norway.xvi+310 pp.http://www.amap.no/documents/doc/amap-assessment-2002-persistent-organic-pollutants-in-the-arctic/96.
11 Harner T,et al.Physicochemical Properties of Persistent Organic Pollutants.Canadian Arctic Contaminants Aeeseement ReportⅡ,Volume 5:Sources,Occurrences,Trends and Pathways in the Physical Environment;Northern Contaminants Program:Hull,Quebec.2003:45-49.http://caid.ca/Vol5_Arct_Cont_2003.html.
12 Gouin T,Mackay D,Webster E,et al.Screening chemicals for persistence in the environment.Environmental Science&Technology,2000,34(5):881—884.
13 Jantunen LM,Bidleman T F.Henry’s law constants for hexachlorobenzene,p,p′-DDE and componentsof technical chlordane and estimatesof gas exchange for Lake Ontario.Chemosphere,2006,62(10):1689—1696.
14 Mackay D,Shiu W Y,Ma K C,et al.Handbook of Physical-Chemical Properties and Environmental Fate for Organic Chemicals.2nd ed.West Palm Beach,Florida:CRC Press,2006.
15 Ma JM,Hung H,Tian CG,etal.Revolatilization of persistentorganic pollutants in the Arctic induced by climate change.Nature Climate Change,2011,1(5):255—260.
16 Wu X G,Lam JCW,Xia CH,etal.Atmospheric HCH concentrationsover themarine boundary layer from Shanghai,China to the Arctic Ocean:Role of human activity and climate change.Environmental Science&Technology,2010,44(22):8422—8428.
17 Wu X G,Lam JCW,Xia CH,etal.Atmospheric concentrationsof DDTs and chlordanesmeasured from Shanghai,China to the Arctic Ocean during the Third China Arctic Research Expedition in 2008.Atmospheric Environment,2011,45(22):3750—3757.
18 Chernyak SM,Rice C P,McConnell L L.Evidence of currently-used pesticides in air,ice,fog,seawater and surfacemicrolayer in the Bering and Chukchi seas.Marine Pollution Bulletin,1996,32(5):410—419.
19 Wania F,Hoff JT,Jia CQ,etal.The effects of snow and ice on the environmental behaviour of hydrophobic organic chemicals.Environmental Pollution,1998,102(1):25—41.
20 Gioia R,Lohmann R,Dachs J,etal.Polychlorinated biphenyls in air and water of the North Atlantic and Arctic Ocean.Journal of Geophysical Research:Atmospheres,2008,113(D19):doi:10.1029/2007JD009750.
21 Jantunen L M,Helm P A,Kylin H,et al.Hexachlorocyclohexanes(HCHs)in the Canadian archipelago.2.Air-water gas exchange of alpha-and gamma-HCH.Environmental Science&Technology,2008,42(2):465—470.
22 Wong F,Jantunen LM,Pucko M,et al.Air-Water Exchange of Anthropogenic and Natural Organohalogens on International Polar Year(IPY)Expeditions in the Canadian Arctic.Environmental Science&Technology,2011,45(3):876—881.
23 Lakaschus S,Weber K,Wania F,etal.The air-sea equilibrium and time trend of hexachlorocyclohexanes in the Atlantic Ocean between the Arctic and Antarctica.Environmental Science&Technology,2002,36(2):138—145.
24 Gregor D J,GummerW D.Evidence of atmospheric transport and deposition of organochlorine pesticides and polychlorinated biphenyls in Canadian Arctic snow.Environmental Science&Technology,1989,23(5):561—565.
25 Welch H E,Muir D CG,Billeck B N,etal.Brown snow:a long-range transportevent in the Canadian Arctic.Environmental Science&Technology,1991,25(2):280—286.
26 Herbert BM J,Halsall C J,Villa S,et al.Rapid changes in PCB and OC pesticide concentrations in Arctic snow.Environmental Science&Technology,2005,39(9):2998—3005.
27 Kang JH,Son M H,Hur SD,etal.Deposition of organochlorine pesticides into the surface snow of East Antarctica.Science of the Total Environment,2012,433:290—295.
28 Yao ZW,Jiang G B,Xu H Z.Distribution of organochlorine pesticides in seawater of the Bering and Chukchi Sea.Environmental Pollution,2002,116(1):49—56.
29 Muir D,Strachan W.Persistent organic pollutants in seawater.Canadian Arctic Contaminants Assessment ReportⅡ,Volume 5:Sources,Occurrences,Trends and Pathways in the Physical Environment;Northern Contaminants Program:Hull,Quebec.2003:92—99.http://caid.ca/Vol5_Arct_Cont_2003.html.
30 Jantunen L M,Kylin H,Bidleman TF.Air-water gasexchange ofα-hexachlorocyclohexane enantiomers in the South Atlantic Ocean and Antarctica.Deep Sea Research PartⅡ:Topical Studies in Oceanography,2004,51(22-24):2661—2672.
31 Weber J,Halsall C J,Muir D CG,et al.Endosulfan andγ-HCH in the Arctic:An assessment of surface seawater concentrations and air-sea exchange.Environmental Science&Technology,2006,40(24):7570—7576.
32 Li Y F.Global gridded technical hexachlorocyclohexane usage inventories using a global cropland as a surrogate.Journal of Geophysical Research:Atmospheres,1999,104(D19):23785—23797.
33 Xiao H,Li N Q,Wania F.Compilation,evaluation,and selection of physical-chemical property data forα-,β-,andγ-hexachlorocyclohexane.Journal of Chemical and Engineering Data,2004,49(2):173—185.
34 Hargrave B T,Barrie L A,Bidleman T F,etal.Seasonality in exchange of organochlorines between Arctic air and seawater.Environmental Science&Technology,1997,31(11):3258—3266.
35 Bidleman T F,McConnell L L.A review of field experiments to determine air-water gas exchange of persistentorganic pollutants.Science of the Total Environment,1995,159(2-3):101—117.
36 Jantunen L M,Bidleman T F.Reversal of the air-water gas-exchange direction of hexachlorocyclohexanes in the Bering and Chukchiseas:1993 versus1988.Environmental Science&Technology,1995,29(4):1081—1089.
37 Ding X,Wang X M,Xie ZQ,et al.Atmospheric hexachlorocyclohexanes in the North Pacific Ocean and the adjacent Arctic region:Spatial patterns,chiral signatures,and sea-air exchanges.Environmental Science&Technology,2007,41(15):5204—5209.
38 Sahsuvar L,Helm P A,Jantunen LM,et al.Henry’s law constants forα-,β-,andγ-hexachlorocyclohexanes(HCHs)as a function of temperature and revised estimates of gas exchange in Arctic regions.Atmospheric Environment,2003,37(7):983—992.
39 Hung H,Kallenborn R,Breivik K,et al.Atmospheric monitoring of organic pollutants in the Arctic under the Arctic Monitoring and Assessment Programme(AMAP):1993—2006.Science of the Total Environment,2010,408(15):2854—2873.
40 Macdonald RW,Harner T,Fyfe J.Recent climate change in the Arctic and its impacton contaminant pathways and interpretation of temporal trend data.Science of the Total Environment,2005,342(1-3):5—86.
41 Cai M H,Ma Y X,Xie ZY,etal.Distribution and air-sea exchange of organochlorine pesticides in the North Pacific and the Arctic.Journal ofGeophysical Research:Atmospheres,2012,117(D6):doi:10.1029/2011JD016910.
42 Galbán-Malagón C,Berrojalbiz N,Ojeda M J,etal.The oceanic biological pumpmodulates the atmospheric transportof persistentorganic pollutants to the Arctic.Nature Communications,2012,3:862.
43 Hoegh-Guldberg O,Bruno JF.The Impact of Climate Change on theWorld’s Marine Ecosystems.Science,2010,328(5985):1523—1528.
44 Cabrerizo A,Dachs J,BarcelóD,et al.Climatic and Biogeochemical Controls on the Remobilization and Reservoirs of PersistentOrganic Pollutants in Antarctica.Environmental Science&Technology,2013,47(9):4299—4306.
45 Ma JM,Cao Z H.Quantifying the perturbations of persistent organic pollutants induced by climate change.Environmental Science&Technology,2010,44(22):8567—8573.
46 Geisz H N,Dickhut R M,Cochran M A,etal.Melting glaciers:A probable source of DDT to the Antarcticmarine ecosystem.Environmental Science&Technology,2008,42(11):3958—3962.
