总免疫球蛋白E时间分辨荧光免疫分析法的建立与评价*
2014-03-08谭玉华孙宝清
谭玉华,孙宝清
(广州医科大学附属第一医院广州呼吸疾病研究所/呼吸疾病国家重点实验室,广州510120)
免疫球蛋白E(immunoglobulin E,IgE)在血清中含量极微,仅占血清免疫球蛋白总量的0.002%,但它却是引起过敏反应的主要抗体。自1966年日本学者Ishizaka发现IgE以来,开启了过敏性疾病研究的新时代。总IgE(total IgE,TIgE)的测定已在临床广泛应用,它已成为过敏性疾病诊断与鉴别诊断、寄生虫感染性疾病、免疫功能障碍相关性疾病的实验室评估指标。随着环境的改变,在生活中接触到的食物、药物、化学物质等越来越多,过敏原也就越来越复杂多样,可达数千种,而且很隐蔽。虽然目前已有多种国际先进的过敏原检测系统,但仍难将所有过敏原全部涵盖。TIgE检测对这类无法确定具体过敏原的过敏症患者仍具有极大的帮助。TIgE的体外定量检测可为判定机体对过敏原的易感性提供了有价值的信息[1],TIgE是特应性体质的客观指标。据文献报道,血清TIgE水平的高低还与疾病严重程度有关,测定TIgE含量有助于对病情严重程度的判定从而对急性发作期临床治疗疗程、缓解期预防性用药疗程进行指导[2]。IgE在过敏性疾病诊断中的作用已获得公认,但血清中IgE的含量较低,特别是新生儿脐带血中IgE含量极低,必须用灵敏的方法才能正确检测。建立一个高度敏感同时又非常特异的检测系统是开展人IgE抗体免疫调节机理研究的基本前提和难点之一。目前,时间分辨荧光免疫分析(time-resolved fluoroisnmunoassay,TRFIA)已成为生物医学研究和临床超微量生化检验中一项最有发展前景的分析手段。因此,笔者基于双抗体夹心法建立了TIgE TRFIA法,并对其性能进行了评价,现报道如下。
1 材料与方法
1.1 材料 按照《全国临床检验操作规程》(第3版)采集静脉血,样本如其干扰限超过试剂盒说明书给定范围或样本污染等均应予以剔除。收集用于方法学比对试验预期偏倚评估的随机样本共40例,其待测物浓度应覆盖方法学的检测范围,且尽可能均匀分布;健康成人样本来自健康体检正常者的血清样本共20例,用于参考值的转移接收的验证试验;确诊为过敏性疾病患者的样本80例,用于评价TIgE在过敏患者样本中的检出率及方法学检测结果间的一致性。
1.2 仪器与试剂 TAKEEN-Ⅰ型TRFIA仪、FWZ-Ⅰ型微量振荡器、DEM-Ⅲ型全自动洗板机(广州市丰华生物工程有限公司);酶标仪(深圳雷杜生命科学股份有限公司);VictorTM2D1420型TRFIA仪(芬兰Wallac Oy公司)。羊抗人IgE多克隆抗体(深圳菲鹏生物股份有限公司);鼠抗人IgE单克隆抗体(美国Advance公司);人IgE纯品(英国Abcam公司);牛血清清蛋白(BSA)和Tween20(美国Sigma公司);Sepharose CL-6B凝胶(瑞典Pharmacia公司);96孔微孔板(深圳金灿华实业有限公司);铕(Eu3+)标记试剂盒(芬兰 Wallac Oy公司);带有滤膜的离心管(美国Millipore公司);其余试剂为国产分析纯试剂;自制以β-萘甲酰三氟丙酮为主要成分的增强液;自制含0.4%(V/V)的 Tween20为主要成分的 pH 7.8 200 mmol/L Tris-HCl溶液为浓缩洗涤液;自制含10%(V/V)的小牛血清和0.01g/L乙二胺四乙酸二钠为主要成分的pH 7.8 50mmol/L的Tris-HCl溶液为实验缓冲液;自制含50g/L BSA的50mmol/L、pH 7.8的Tris-HCl缓冲液为校准品稀释液或样本稀释液。300mg/mL的IgA、IgG、IgM,160g/L的清蛋白,1 000IU/mL的甲胎蛋白,800IU/mL的类风湿因子,低、中、高3个浓度的质量控制品(广州市丰华生物工程有限公司);IgE国际标准品(WHO 2ndIRP75/502;5 000IU/安培)(英国NIBSC);TIgE检测ELISA试剂盒(德国欧蒙医学实验诊断股份公司)。
1.3 方法
1.3.1 校准品的制备 以 WHO 2ndIRP 75/502为对照,人IgE纯品用含50g/L BSA的50mmol/L、pH 7.8的 Tris-HCl缓冲液定量稀释成0.0、2.4、12.0、60.0、300.0、1 500.0IU/mL(分别用A~F表示)。
1.3.2 固相反应板的制备和最适包被浓度的选择 将羊抗人IgE多克隆抗体用50mmol/L、pH 9.6的碳酸盐缓冲液稀释成包被液(2.0、3.0、4.0、5.0、6.0μg/mL),在固相微孔板加入100μL抗体包被液,以上浓度各包被1块,4℃冰箱孵育过夜,洗涤1次,再每孔中加入含10g/L BSA的50mmol/L、pH 7.2的磷酸盐缓冲液(PBS)250μL,37℃恒温箱孵育2h封闭,甩干后晾干,真空封膜,4℃冰箱保存备用。采用方阵(棋盘)滴定法,在2.0~6.0μg/mL预包被的反应板上,检测校准品A、校准品F 2个浓度点的荧光值强度(CPs)。选取校准品A本底荧光值(N)较低,且校准品F的检测荧光值(P)与N的比值(P/N)最大的包被浓度为最适包被浓度。确定最适包被浓度后按以上方法制备固相反应板。
1.3.3 铕标记物的制备和最佳稀释度的选择 将1mg的鼠抗人IgE单克隆抗体加入Millipore公司的带有滤膜的离心管中,10 000r/min离心9~10min。再用标记缓冲液(50mmol/L、pH 9.3的碳酸盐缓冲液)重复洗涤3~5次。将收集到的200μL鼠抗人IgE单克隆抗体和1mg的铕标记试剂充分混匀,4℃振荡反应(24±2)h。反应液经50mmol/L、pH 7.80 Tris-HCl缓冲液平衡的Sepharose CL-6B柱(1cm×30cm)层析,每管2.0mL,A280监测收集第一洗脱峰。合并峰管,根据Eu3+标记试剂盒说明书所提供的公式计算标记率和回收率。蛋白相对分子质量为160 000的单克隆抗体的理想标记Eu3+个数为6~10个/蛋白分子。将铕标记物用实验缓冲液按体积比1∶500、1∶1 000、1∶1 500、1∶2 000稀释,检测校准品 A、校准品F 2个浓度点的CPs。选取N较低,且P/N最大的稀释度为铕标记物最佳稀释度。
1.3.4 TIgE TRFIA方法学的建立 为了尽量避免 HOOK效应,选用双抗体夹心二步法。检测方法应适用于普通实验室条件下,反应宜选择室温20~25℃条件。第1步:在固相微孔反应板中依次加入10μL的校准品或样本或质量控制品,再每孔中加入100μL样本稀释液,在室温条件下振荡孵育(30、45、60、75min),洗板5次拍干。第2步:在每孔中加入100μL铕标记物工作液,在室温条件下振荡孵育(30、45、60、75min),洗板5次拍干。然后在每孔中加入100μL增强液,在室温条件下振荡(1、2、3、4、5、10、30、40min),将反应板放入 TRFIA 仪内进行荧光计数。选择检测校准品A~F荧光值接近平衡的反应时间作为最适反应时间。
1.3.5 性能评价 参考文献[3-4]对剂量-反应曲线拟合模型进行优选。参考NCCLS的指南及有关文献[5-14]对精密度、检测低限、线性、准确度(校准品核对试验、回收试验)、特异性(交叉反应试验、干扰试验)、参考值确认、方法学比对等进行评价。
1.4 统计学处理 TRFIA测量所得荧光计数采用TRFIA仪上随机配备的分析软件进行数据分析。其他统计数据采用SPSS13.0和Microsoft Excel软件进行分析。
2 结 果
2.1 铕标记鼠抗人IgE抗体的鉴定 铕标记鼠抗人IgE抗体A280监测收集,收集铕标记物峰(5~8管)。铕标记鼠抗人IgE抗体的回收率为92.51%,表明分离纯化后的抗体损失较少。以芬兰Wallac Oy公司标准Eu3+为参考,铕标记物峰的铕标记鼠抗人IgE抗体中的Eu3+含量为6.06μmol/L,蛋白含量为0.72μmol/L,计算可得平均每个鼠抗人IgE抗体上连接了8.40个Eu3+,达到了理想的标记率。
2.2 方法学的建立
2.2.1 最适包被浓度和铕标记物最佳稀释度 经方阵(棋盘)滴定法,当以4.0μg/mL包被反应板和铕标记物按1∶1 500稀释时,本底良好(<2 000)且校准品F检测荧光值的S/N值趋于最大,因此最适包被浓度为4.0μg/mL,铕标记物最佳稀释度为1∶1 500。
2.2.2 反应动力学 当第1、2步的反应时间分别为60min和45min,检测校准品A~F的荧光值趋于“饱和”,曲线斜率趋于最大,表明反应趋于完全。解离增强振荡5min时,校准品A~F的荧光值已趋于平衡,表明Eu3+已从铕标记物螯合物上充分解离下来,与增强液成分形成了稳定的复合物。
2.2.3 建立剂量-反应曲线 按优化的反应条件进行试验[3-4],剂量-反应曲线拟合优选Log_LogB双对数数学模式及三样条平滑(SPLINE)拟合时曲线平滑,拟合程度高,拟合后的曲线的r>0.990 0,典型的剂量-反应曲线,见图1。

图1 TIgE TRFIA的剂量-反应曲线
2.3 分析性能评价
2.3.1 精密度 参考 NCCLS EP5-A文件[5]评价,低、中、高3个浓度的质量控制品批内和批间检测的实测值分别为(4.03±0.07)IU/mL和(4.03±0.30)IU/mL,(90.57±1.44)IU/mL和(90.57±5.05)IU/mL,(319.55±5.33)IU/mL 和(319.55±16.70)IU/mL,批内和批间的变异系数(CV)分别为1.68%和7.33%,1.59%和5.57%,1.67%和5.23%。
2.3.2 检测低限 参考文献[6]评价,平行检测校准品A 20次的荧光值(±s)为1 309+156;95%CI(x±2s)为1 620,在剂量-反应曲线上拟合得到的浓度值为0.25IU/mL,优于欧蒙TIgE ELISA法试剂的检测低限1IU/mL。
2.3.3 线性 参考 EP6-A 文件[7]评价,该方法在1.47~1 510.00IU/mL范围内,其线性回归方程为Y=0.988 2X-0.068 8,r=0.999 3(tr=83.15,v=9,P<0.01),见图2。本方法的线性上限高于欧蒙TIgE ELISA法试剂的线性上限500 IU/mL。
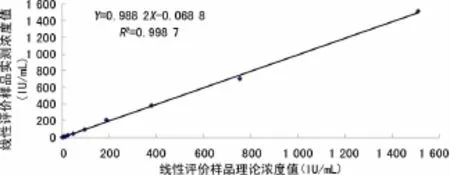
图2 TIgE TRFIA的线性
2.3.4 校准品核对试验 参考文献[8-9]评价,以国际标准品的稀释溶液(2.4~1 500.0IU/mL)为对照,测定TIgE TRFIA各校准品,以相对偏倚±10.00%为可接受范围;TIgE TRFIA校准品A~F的实测值与标示值的相对偏倚在-5.57%~7.92%内。
2.3.5 回收试验 将1份22IU/mL的常规检测样本分成3等份(各1mL),在其中2份中分别加入44IU/mL和110IU/mL的血清样本0.1mL,制成加入浓度分别为4.0IU/mL和10.0IU/mL的2份待回收分析样本,第3份中加入校准品稀释液0.1mL制成基础样本血清,回收率在90.00%~110.00%范围内为可接受。检测基础样本和2份待回收分析样本的实测浓度分别为19.41、24.21IU/mL和29.64IU/mL,2份待回收分析样本的回收率分别为97.00%和106.75%,平均回收率为101.88%,比例系统误差为1.88%。

图3 血红蛋白对样本检测结果的干扰程度
2.3.6 交叉反应 参考文献[9-10]评价,检测300mg/mL的IgA、IgG、IgM,160g/L的清蛋白,800IU/mL的类风湿因子,1 000IU/mL的甲胎蛋白,其实测浓度值分别为0.075、0.120、0.120、0.105、0.125IU/mL和0.040IU/mL,均未高于检测低限0.25IU/mL,表明对TIgE TRFIA检测结果无明显交叉反应。
2.3.7 干扰试验 参考 NCCLS EP7-A2文件[11]评价,分别选用低、中、高浓度的血清加入以下干扰物,干扰物质溶液与血清分别按体积比1∶9混合,组成系列干扰物浓度,对照血清加入干扰物相同体积的纯化水,采用干扰率为±10.00%作为系统干扰物最高限。样本中含血红蛋白(≤18.00g/L),胆红素(≤818.0mol/L),三酰甘油(≤21.54mmol/L),乙二胺四乙酸钾(≤2.20mg/mL),草酸钾(≤2.00mg/mL),肝素(≤15.00U/mL),枸橼酸钠(≤21.80mmol/L),氟化钠(≤1.00mg/mL)对检测结果干扰率均在±10.00%以内,见图3~10。

图4 胆红素对样本检测结果的干扰程度
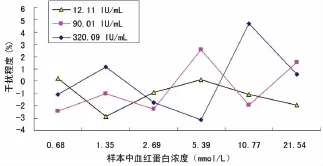
图5 三酰甘油对样本检测结果的干扰程度
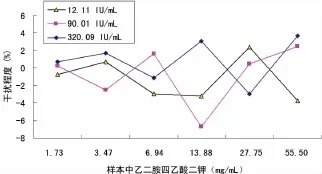
图6 乙二胺四乙酸二钾对样本检测结果的干扰程度

图7 草酸钾对样本检测结果的干扰程度
2.3.8 HOOK效应 将已知浓度的IgE纯品稀释成3.6~15 000.0IU/mL的系列浓度样品,每稀释浓度重复测定2次后取平均值。该方法检测TIgE达15 000.0IU/mL时仍未见HOOK效应。
2.3.9 参考值验证 欧蒙ELISA法试剂的参考值为100IU/mL(年龄大于16岁)。参考NCCLS C28-A2文件[12]及 WS/T 402-2012[13],对参考值进行转移接收验证,检测20例健康成人样本,仅有1例结果(114.96IU/mL)高于100IU/mL,其他19例样本在29.81~92.91IU/mL间,检测值在引用参考区间的参考个体数与总的参考个体数的比率为95%,可以直接引用100IU/mL作为TRFIA检测成人样本TIgE的参考值,但是健康人群TIgE水平易受环境、种族、遗传、年龄、检测方法及取样标准等因素的影响,各实验室应建立自己的参考值或参考值验证。

图8 肝素对样本检测结果的干扰程度

图9 枸橼酸钠对样本检测结果的干扰程度

图10 氟化钠对样本检测结果的干扰程度
2.3.10 方法学比对 参考NCCLS EP9-A2文件[14]进行预期偏倚评估试验。参考美国CLIA′88规定的类似项目(IgG)的允许误差(±25%)的1/2(即±12.50%),作为方法学可比性的临床接受判断标准。TIgE TRFIA与欧蒙ELISA平行检测40份样本(14.43~518.81IU/mL)结果的线性回归方程为Y=1.005 6X-0.153 4,r=0.999 2(tr=153.99,v=38,P<0.01)。选用欧蒙TIgE ELISA说明书上的各年龄阶段的参考值作为给定值,TRFIA与欧蒙ELISA的测定结果的相对偏倚在-12.22%~0.48%内,均在可接受范围(±12.50%)之内。2种方法在80例过敏性疾病患者样本中均检出61例(76.25%)TIgE高于临界值100IU/mL,2种方法检测结果的一致性符合率为100%。
3 讨 论
目前应用于TIgE检测的方法有免疫比浊法(ITM)、放射免疫法(RIA)、ELISA法、化学发光法(CLIA)、电化学发光法(ECLIA)、荧光免疫分析法(FIA)等。ITM是一类均相免疫测定法,但单纯就灵敏度而言,RIA、ELISA及FIA法等均优于ITM法。因RIA具有放射性污染的固有缺陷,其市场占有率逐年下降,Gosling[15]在《Clinical Chemistry》发表的文章统计,国际上在免疫分析中采用同位素标记所占的比例由1980年的60%下降为1989年的25%,非同位素标记由1980年的不足40%上升为1989年的75%,近年来这个比例仍在变化。目前,临床检测TIgE常用的仍是ELISA法,但其灵敏度较低,线性范围较窄,结果易受操作人员、实验条件等因素的影响。由于一般FIA法中的本底较高等问题,传统的FIA用于定量测定有一定困难。CLIA和ECLIA是目前的新型超微量免疫分析技术,但是CLIA发光过程短、样品不能重复检测、本底高及易受环境物质干扰。ECLIA尽管有一定优势,但目前仪器和试剂均依赖进口,且非开放系统,造成试剂价格昂贵,仪器成本及维护费用高。近年来引进的荧光酶免疫法Pharmacia CAP变应原体外检测系统,试剂价格昂贵,限制了其大量使用。
RIA、ELISA、传统的FIA均没有实现人们所期待的高灵敏度分析,因此科学家们努力寻求一种更可靠的标记物,自1979年Soini等[16]合成了一种可用于标记抗体的“Eu3+、β-二酮体和EDTA衍生物”的三元混合物作为荧光探针并提出TRFIA理论,他们预言TRFIA将发展为新一代以镧系元素为示踪物和时间分辨荧光测量相结合的非放射性微量免疫分析技术。TRFIA因镧系元素的荧光寿命可达60~900s,甚至更长,比普通荧光高数个数量级,在每个激发光脉冲过后,通过延时,门宽选择,并通过滤光,以达到消除来自样品、试剂及其他的非特异荧光,从而提高灵敏度,又因镧系元素的激发光和发射光之间有一个较大的stokes位移,这有利于排除其他非特异荧光的干扰,方法的特异性较高。TRFIA将抗原抗体反应与荧光物质发光和时间分辨技术三者相结合,其灵敏度可达10-18mol/L。与现在广泛使用的 RIA、ELISA、FIA、CLIA和ECLIA相比,TRFIA克服了RIA放射性污染的不足、酶标记物的不稳定性和定量范围窄的缺陷、化学发光仅能一次发光、一般荧光标记受环境干扰的难点和电化学发光的非直接标记等缺点。
本研究应用Eu3+标记建立的TIgE双抗体夹心TRFIA,经方法学评价具有以下优点:(1)重复性好;(2)灵敏度高;(3)线性范围宽;(4)准确度高;(5)特异性强;(6)与欧蒙 ELISA 比对检测试剂线性范围内的临床样本具有良好的相关性,2种方法检测结果的一致性符合率为100.00%;在过敏性疾病患者中的TIgE的阳性检出率达76.25%。表明TIgE的检测在过敏性疾病筛选试验中是一个重要参考指标,本研究建立的TIgE TRFIA能满足临床的需要。
[1] 李丰,曾华松.IgE介导的儿童过敏性疾病的诊断及治疗[J].临床儿科杂志,2013,31(1):97-98.
[2] Lee JH,Haselkorn T,Chipps BE,et al.Gender differences in IgE-mediated allergic asthma in the epidemiology and natural history of asthma:Outcomes and Treatment Regimens(TENOR)study[J].J Asthma,2006,43(3):179-184.
[3] 乐爱平,胡国信,李太原,等.血清乙型肝炎病毒大蛋白浓度标准曲线的拟合与优选[J].检验医学,2009,24(6):463-465.
[4] 胡镇球,夏宗勤.免疫放射分析剂量效应曲线的质量作用模型及其拟合[J].中华核医学杂志,1992,12(4):226-228.
[5] National Committee for Clinical Laboratory Standards.EP5-A Evaluation of precision performance of clinical chemistry devices,approved guideline[S].Wayne,PA:NCCLS,1999.
[6] 冯仁丰.分析灵敏度 (检测限)[J].上海医学检验杂志,2002,17(3):133-136.
[7] National Committee for Clinical Laboratory Standards.EP6-A Evaluation of the Linearity of Quantitative measurement Procedures:A Statistical Approach,Proposed Guideline[S].Wayne,PA:NCCLS,2003.
[8] 国家食品药品监督管理局.YY/T 1175-2010中华人民共和国医药行业标准——肿瘤标志物定量测定试剂(盒)(化学发光免疫分析法)[S].北京:中国标准出版社,2012.
[9] 徐伟文.体外诊断试剂研制常用技术指标之分析性能评估[J].分子诊断与治疗杂志,2010,2(2):140-144.
[10] 董劲春.过敏原特异性IgE抗体检测试剂盒的临床研究指南[J].标记免疫分析与临床,2011,18(3):213-216.
[11] National Committee for Clinical Laboratory Standards.EP7-A2Interference testing in clinical chemisty,approved guideline-second edition[S].2nd.Wayne,PA:NCCLS,2005.
[12] National Committee for Clinical Laboratory Standards.C28-A How to Define and Determine Reference Intervals in The Clinical Laboratory,Approved Guideline [S].Wayne,PA:NCCLS,2000.
[13] 中华人民共和国卫生部.WS/T 402-2012中华人民共和国卫生行业标准——临床实验室检验项目参考区间的制定[S].北京:中国标准出版社,2013.
[14] National Committee for Clinical Laboratory Standards.EP9-A2Method comparison and bias estimation using patient samples,approved guideline[S].2nd.Wayne,PA:NCCLS,2002.
[15] Gosling JP.A decade of development in immunoassay methodology[J].Clin Chem,1990,36(8):1408-1427.
[16] Soini E,HemmiläI.Fluoroimmunoassay:present status and key problems[J].Clin Chem,1979,25(3):353-361.
