High-throughput Sequencing Technology and Its Application
2014-03-07ZhuQianglongLiuShiGaoPengandLuanFeishi
Zhu Qiang-long, Liu Shi, Gao Peng, and Luan Fei-shi
College of Horticulture, Northeast Agricultural University, Harbin 150030, China
High-throughput Sequencing Technology and Its Application
Zhu Qiang-long, Liu Shi, Gao Peng, and Luan Fei-shi*
College of Horticulture, Northeast Agricultural University, Harbin 150030, China
Gene sequencing is a great way to interpret life, and high-throughput sequencing technology is a revolutionary technological innovation in gene sequencing researches. This technology is characterized by low cost and high-throughput data. Currently, high-throughput sequencing technology has been widely applied in multi-level researches on genomics, transcriptomics and epigenomics. And it has fundamentally changed the way we approach problems in basic and translational researches and created many new possibilities. This paper presented a general description of high-throughput sequencing technology and a comprehensive review of its application with plain, concisely and precisely. In order to help researchers finish their work faster and better, promote science amateurs and understand it easier and better.
high-throughput sequencing, data analysis, genome sequence, transcriptome sequence, bioinformatics
Introduction
With the indepth study of life sciences and further development of bio-technology, more and more scientists recognize that the whole genome sequencing of a species will be the fundamental basis and important clue to help them reveal the nature of life of the species. The discovery of DNA double helix (Watson and Crick, 1953), cracking genetic code (Nirenberg et al., 1966), and the successful completion of the first one complete genome map (Sanger et al., 1977) have undoubtedly become a series of important journey milestones in the history of life scientific development, and make more scientists profoundly recognize that sequencing technology plays an important role in life science researches. The rapid sequencing technology would make DNA sequencing become one of the most important methods of molecular analysis (Sanger et al., 1992). This technology provides important data for basic biology study, such as disclosure of genetic information and regulation of gene expressions. With the appearence of Roche's 454 technology (2005), Illumina's Solexa technology (2006) and ABI's SOLiD technology (2007), high-throughput sequencing technology has got enormous developments, thus amounts of genetic information is successively revealed, which allow us to explore the essence of life in detail, to uncover the huge diversity of novel genes that are currently inaccessible, to understand nucleic acid therapeutics, to better integrate biological information for a complete picture of health and disease at a personalized level and to move to advance that we can not yet imagine (Kahvejian et al., 2008). Therefore, a number of bioinformatics methods and softwares havebeen created to accelerate high-throughput sequencing technology to be widely applied in aspects of genomics researches on genomics, transcriptomics and epigenetics. High-throughput sequencing technology has fundamentally changed the way we approached problems in basic and translational researches and created many new possibilities. Whereas, it has also brought new challenges for bioinformatics: how to effectively process and analyze these massive data and extract valuable bio-information form it, which have become an important key to decide if high-throughput sequencing technology plays a major role in the scientific exploration. In this article, we intended to present a comprehensive and systematic introduction of high-throughput sequencing technology and its applications to the enthusiast of biological science with plain, concisely and precisely hope to help researchers finish their work faster and better, to promote science amateurs understand it easier and better. Meanwhile, we tried to take data generated from Illumina Hiseq 2000 sequencing platform as an example to present a more complete description of the basic procedure, key methods and existing software of the sequencing data generating process, data processing and analysis.
History of High-throughput Sequencing Technology Development
High-throughput sequencing technology is the second generation sequencing technology launched by Roche/454 Company, Illumina/Solexa Company and ABI/SOLiD Company based on Sanger sequencing and single-molecule sequencing technologies announced by Helicos HeliscopeTMand Pacific Biosciences, which is also called as deep sequencing technology (Sultan et al., 2008) or the next-generation sequencing technology (NGS) (Schuster, 2008) .
The 1st generation sequencing technology
In 1977, Sanger of Cambridge and Gilbert of Harvard almost simultaneously published their different methods of DNA sequencing in the same magazine (Maxam and Gilbert, 1977; Sanger et al., 1977), their inventions first opened a door to study the genetic code of life deeply for researchers, and brought hope to the development of faster and more efficient sequencing technology. Sanger method belongs to dideoxy chain termination method, while Gilbert method is chemical degradation method. The former is more convenient and more suitable for optical automatic detection gradually replaced the latter, and became the most widely applied method of sequencing in the field of life science. Thus, Sanger won the 1980 Nobel Prize in chemistry (Sanger, 1988). Most of the automated DNA sequencers are based on this method. Its principle is as below, when a nucleic acid template is replicating under the presence of DNA polymerase, a pair of primers, four types of single deoxynucleotide triphosphate (dNTP, one of them labeled with a radioactive 32P), join four kinds of dideoxynucleotide triphosphate (ddNTP) into four reactive systems in proportion, because dideoxynucleotide have no 3'-OH, so long as the dideoxynucleotide append to the end of the chain, its extension is stopped, if the single deoxynucleotide triphosphate append to the end of the chain, it can continue to be extended. So that a series of the nucleic acid fragments with the dideoxy nucleotide at the 3' end in different length ranges will be synthesized in each reaction system. After termination of the reaction, different lengths of nucleic acid fragments should be isolated by gel electrophoresis in four lanes, where there is a difference of one nucleotide among near segments. After autoradiography, the order of base in synthetic fragment can be read, according to the dideoxy nucleosides at the 3' end of the fragment (Xie et al., 2010). Subsequently, a variety of DNA sequencing technologies based on this technology has been exploited, the most important one of them is fluorescent automated sequencing technology (Fig. 1) (http:// en.wikipedia.org/wiki/File:Sanger-sequencing.svg). This generation sequencing technology has played a key role in human genome project, accelerating the completion of human genome project. The sequencerusing this technology still be used in today, which will continue to play an important role, because it has an obvious advantage in the original data quality and read length, and has been used widely in different fields, especially in PCR products sequencing, plasmids and bacterial artificial chromosomes terminal sequencing, and Short Tandem Repeat (STR) genotyping (Zhou et al., 2010). The dependence on electrophoretic separation, however, makes it difficult to further enhance the speed of analysis and to reduce sequencing cost by mini-aturization. Therefore, developing new technologies to break these limitations is needed.
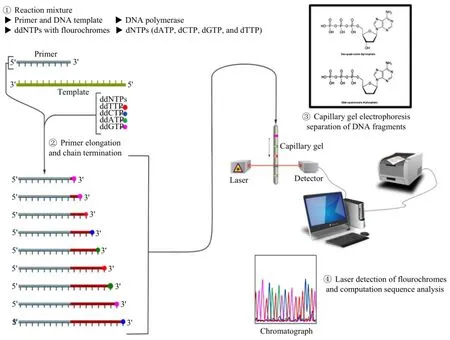
Fig. 1 Sanger (chain-termination) method for DNA sequencing
The second generation sequencing technology
Compared with the Sanger sequencing method, the second generation sequencing technology is also called as next-generation sequencing technology. The second generation sequencing technology are mainly classified into three major sequencing techniques: Roche/454 pyrosequencing (2005), Illumina/Solexa sequencing of polymerase synthesis (2006) and ABI/SOLiD sequencing ligase (2007) technology. Compared with Sanger sequencing, the common prominent feature in three kinds of next-generation sequencing technologies is that they could output massive data in a single run, thus they are also known as high-throughput sequencing technologies (Ansorge, 2009). And their core idea is sequencing-by-synthesis. When generating a new complementary strand of cDNA, they eitheradded normal dNTP through enzymatic cascade reaction to catalyze substrates to excite fluorescence (Roche/454), or directly into the fluorescently labeled dNTP (Illumina/Solexa) or semi-degenerate primers (ABI/SOLiD), then when generating or connecting to synthesize the complementary chain, the substrates will release fluorescent signal. By capturing the optical signal and converting to a sequencing peak, it can be converted again to the sequence information of complementary strand. High-throughput sequencing technology achieved massively parallel sequencing (MPSS), so the cost of getting a base data declined lower than Sanger method, and it has been applied in multi-level researches on medical science, agriculture science and life science (Aksyonov et al., 2006).
The third generation sequencing technology
Although the second generation sequencing technology, compared with the first generation sequencing technology, has greatly improved and been more widely used in many aspects, but still built on the basis of PCR amplification. In order to reduce deviation and cost caused by PCR amplification, scientists are now developing the third generation sequencing that directly sequence a single molecule of DNA. The most representative technologies included Heliscope single-molecule sequencing, single molecule realtime compositing sequencing, nanopore sequencing technology. Helicos was a sequencing technology, based on total internal reflection microscopy (TIRM)—single-molecule sequencing technology. The technology completely gave up the signal amplification process of sequencing platform based on PCR amplification, but was still based on sequencingby-synthesis principle (Harris et al., 2008), which used a new fluorescent analogs and sensitive monitoring system that would be directly capable of recording fluorescent form a single nucleotide, thereby overcoming the defect of other methods that need to simultaneously test thousands of the same genes to increase the signal intensity. Soon after, PacificBiosciences developed another single molecule sequencing technology-single molecule real-time technology (SMRT) (Eid et al., 2009). The sequencing technology take full advantages of DNA polymerase, which can be vividly described as a real-time observation on DNA polymerase through the microscope. In a word, it records the entire process of DNA synthesis. Nanopore sequencing technique (Rusk, 2009) was to use the subtle changes of electrostatic induction caused by different bases passing the nanopore to identify the types of the base signal. Meanwhile, it could detect some important information, for example, whether a base was being methylation or not.
Main Methods and Steps of Highthroughput Data Analyses
The data generated from Illumina Hiseq 2000 (Fig. 2) (http://bitesizebio.com/13546/sequencing-by-synthesis-explaining-the-illumina-sequencing-technology/) sequencing platform was taken to present a more complete description of the basic procedure, key methods and existing software of the sequencing data generating process, data processing and analysis.
Statistics and filtering of raw sequence data
Through the base calling, the original image data can be transformed into sequence data, which is called raw data or raw reads, which are usually storied in a file with the format of fastq and is the most original file that users would get, which stores not only the sequence of reads, but also quality of sequencing reads. Each read in the fastq file is described by four lines:
@ WATERMALON: 1:8:6:490
CCACTGTCATGTGAACATCACAGAGACATT TCTTGA
+
bbbbbbbbbbbbbbbbbbbbbbbbbaaaaaaaaa_
Lines 1 and 3 are sequence names generated by the sequencing machines; line 2 is sequence; line 4 is quality letter, of which each letter corresponds to a base in line 2; we calculate the sequencing quality ofeach base in line 2 by subtracting 64 from ASCII value of the letter in line 4 (sequencing quality value). For example, ASCII value of c is 99, so the corresponding sequencing quality value is 35. Sequencing quality values range from 2 to 35. After data is outputted, there should be a statistics on reads obtained from the sample sequencing, the length of reads per sample, the number of nucleotides, GC content and so on, which helps to assess whether the quality of data meets the requirements to analyze. Then, the original data still need some basic pre-processing according to the result. For example, removing reads with adaptor, removing to reads with N ratio greater than 5%, removing low quality reads (the number of base with Q≤20 is 50% or more of the total number of bases) to obtain clean reads for the further analyses.
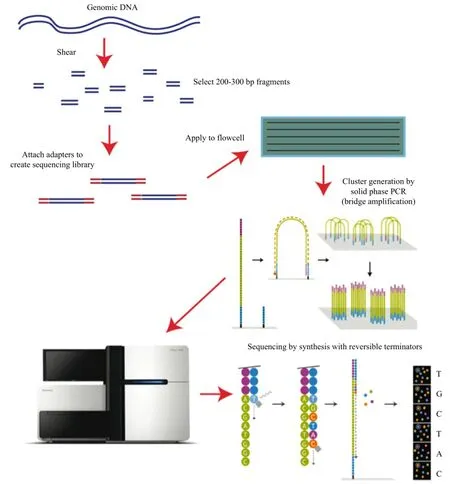
Fig. 2 Sequencing method of Illumina Hiseq 2000
Data assembly and mapping
It mainly contains re-sequencing with a reference genome to locate reads and de novo genome sequencing assembly without reference. Re-sequencing read paragraphs orientation: it refers to data assembly with reference genome. When raw data generated, firstly, all reads should be sorted by their length, then mapped to the reference genome, and analyzed them through comparing with reference genome to pick out all goodmatch reads, which is important for all subsequent processing and analysis. Re-assembly has been widely applied in the model plant with reference genome (Birol et al., 2009; Cheung et al., 2006), mostly softwares used to assembly are: BWA (Li and Durbin, 2009), SOAP2 (Li et al., 2009), Bowtie (Langmead et al., 2009), MAQ (Li et al., 2008), ZOOM (Lin et al., 2008), TopHat and cufflinks (Trapnell et al., 2012) etc. De novo sequencing assembly: first the sequencing reads will be orderly assembled into contig, then the contig will be assembled into the scaffold, and use N to fill with the intermediate gap. Finally get the sequence without N, which cannot be extended at both ends, called unigene. And blastx alignment (evalue<0.00001) between unigenes and protein databases like Nonredundant (Nr), UniProt Knowledgebase (UniProtKB), Kyoto Encyclopedia of Genes and Genomes (KEGG) and Cluster of Orthologous Group (COG) is performed, and the best aligning results are used to decide sequence orientation of unigenes. If results of different databases conflict with each other, a priority order of Nr, UniProtKB, KEGG and COG should be followed. When a unigene happens to be unaligned to none of the above databases, a software named ESTScan (Iseli et al., 1999) will be introduced to decide its sequence direction. De novo assembly provides an efficient way to quickly obtain expressive genes from the short sequence and no reference sequence of assembly. Due to longer reads of Roche/454 technology, so it was more suitable for assembly, while it was considerable difficult for Illumina and SOLID technology in the splicing strategy how to splice the short length reads into a long sequence because of the short read (Weber et al., 2007). In recent years, researchers have designed a variety of softwares to solve the problems to make data from Illumina more suitable for assembly, and achieved good effects of splicing. The technology that contains three kinds of sequencing platforms has been widely applied in a lot of non-model animals and plants (Butler et al., 2008). Currently, the most commonly used softwares are: Trinity (Grabherr et al., 2011), SOAP denovo, Velvet ( http://www.ebi.ac.uk/-zerbino/velvet/), etc.
ldentification and functional annotation of genes
Currently, the main principles of gene identification and functional annotation are the followings: (1) sequence searching. Its hypothesis: sequence similarity=homology=similar function. (2) Sequence motif. In case of no significant sequence homology, it's used to find the local features of sequence. (3) COGs of proteins. (4) Subcellular localization. It's to predict function of gene by predicting subcellular localization. (5) Structure comparison. First predict unknown gene protein structure, then predict its functions through structure comparison. (6) Proteomics. To predict protein function by the networks of protein interaction, or other biomolecules networks. Sequence searching that based on the assumption "homologous equal functionality similar" is widely used, most of websites and softwares for annotating gene function are primarily based on this principle at present. They take full advantages of bioinformatics methods to speculate the unknown genes' function, making the unknown gene sequences (e.g. unigene) search against public database, then obtain the highest similar annotated sequences with the given-query sequences. Main annotated nucleotide databases are: GenBank (NCBI), EMBL, DDBJ, etc., protein databases are: Nr, UNIPROTKB, TrEMBL, COG, etc. Main comparing software: BLAST, FASTA, etc. There are mainly two ways to annotate the currently gene function: Gene Ontology (GO) classification and KEGG func-tional classification. GO is an international standardized gene functional classification system which offers a dynamic-updated controlled vocabulary and a strictly defined concept to comprehensively describe properties of genes and their products in any organisms. GO has three ontologies: molecular function, cellular component and biological process, which is applicable to define and describe gene function of any species (Ashburner et al., 2000). KEGG is a database that is able to anaylze gene product in metabolism process and related gene function in the cellular processes. With the help of KEGG database, we can further study genes' biological complex behaviors (Altermann and Klaenhammer, 2005). Softwares that are mostly used including: Blast2go (Conesa et al., 2005), WEGO (Ye et al., 2006), GoMiner (Zeeberg et al., 2003), DAVIA (Dennis et al., 2003), VisANT (Hu et al., 2009) etc.
Application of High-throughput Sequencing Technology
It could be argued that the greatest transformative aspect of the Human Genome Project has not been the sequencing of the genome itself, but the resultant development of new technologies. Since 454 Company developed the first full-automatic sequencer to open the prelude to a new era of high-throughput DNA sequencing in 2005, the technology had achieved a leap-type development, and brought genomics level research into a new era. Meanwhile, this technology has already made molecular biologists increase their basic knowledge of genomics into a higher level. Kahvejian et al. (2008) mentioned that high-throughput sequencing technology would allow us to move to advances that we couldn't imagine yet.
DNA level application
Full genome sequencing
Full genome sequencing, also known as de novo sequencing, directly sequence the whole genome of species and then get its complete genome sequences by bioinformatics methods to splice and assemble sequence. The technology has a very important significance for a comprehensive understanding of the molecular evolution of a species, and its gene component and regulation. The sequencing technology had greatly promoted the whole-genome sequencing work of non-model species, and helped scientists free from the obstacles that the non-model organisms had relatively poor genetic background and few base researches on their genes in the past. Full genome sequencing has been applied in multi-level researching areas, especially in the biology. Biologists could make the best of this technology to sequence the genome of important species, which would help them to know the information of gene sequence, to elucidate the evolutionary of the species and to better understand the molecular mechanisms of life. Watermelon (Citrullus lanatus) is an important cucurbit crop grown throughout the world, Guo used high-throughput sequencing technology and Sanger method to complete the watermelon genome sequencing (Guo et al., 2013), and they reported a high-quality draft genome sequence of the east Asia watermelon cultivar 97103 (2n=2×=22) containing 23 440 predicted protein-coding genes. Comparative genomics analysis provided an evolutionary scenario for the origin of the 11 watermelon chromosomes derived from a 7-chromosome paleohexaploid eudicot ancestor. Resequencing of 20 watermelon accessions representing three different C. lanatus subspecies produced numerous haplotypes and identified the extent of the genetic diversity and population structure of watermelon germplasm. Genomic regions that were preferentially selected during domestication were identified. Many disease resistance genes were also found to be lost during domestication. In addition, integrative genomic and transcriptomic analyses gave important insights into aspects of phloem-based vascular signaling in common between watermelon and cucumber and identified genes crucial to valuable fruit-quality traits, including sugar accumulation and citrulline metabolism. Meanwhile, genomicinformation of more and more species have been published, such as potato (Solanum tuberosum) (Xu et al., 2011), Chinese cabbage (Brassica rapa) (Wang et al., 2011), apple (Malus domestica Borkh) (Velasco et al., 2010), and cucumber (Cucumissativus L.) (Huang et al., 2009). In addition, by virtue of its sensitive catch-capability for trace DNA, high-throughput sequencing technology has also been widely used in paleontology researches. Rasmusse et al. (2010) extracted DNA from a bunch of hair of Eskimos 4 000 years ago, then sequenced its whole-genome, gotten about 79% of its sequence and compared with the modern human genome sequence, which provided important information for exploring human evolutionary history.
The whole genome re-sequencing
April 17, 2008, U.S. scientists in Nature published the genome sequencing results of "DNA Father" James D.Watson, which is the first whole genome resequencing results through high-throughput sequencing technology (Wheeler et al., 2008). The whole genome re-sequencing is to sequence different individual genomes of the same species under the condition of knowing its reference genome, and then conduct differential analysis among individuals or groups. Currently, re-sequencing with reference genome is applied widely in the field of the second generation sequencing technology and is also rapidly becoming one of effective methods of breeding and has great scientific values in the whole genome for scanning and detecting important traits of plants and animals associated with mutation sites. By re-sequencing, scientists in the field of agriculture can obtain a lot of Single Nucleotide Polymorphisms (SNPs), Insertions/Deletions (InDels), Structure Variations (SVs), and group's polymorphism. Zheng et al. (2011) carried out the whole genome re-sequencing for three lines of sorghum bicolor with sequencing depth of 12 times, then they took American grain sorghum genome sequence as a reference to conduct information analysis. They uncovered 1 057 018 SNPs, 99 948 InDels of 1-10 bp in length, 16 487 Presence/Absence Variations (PAVs) and 17 111 Copy Number Variations (CNVs). Meanwhile, they identified a cluster of nearly 1 500 genes with structural differences in sweet sorghum and sorghum grain. These genes were involved in metabolisms of sugar and starch, synthesis of lignin and coumarin, nucleic acid metabolism, stress response, biological processes and DNA repair. In addition, in the field of evolution, scientists can apply population polymorphism analysis to explore evolutionary model in different species; in the field of microbiology, DNA sequencing genotyping has been proven faster and more accurate; in the medical field, the genome re-sequencing has important significance in the discovery of relationship between SNPs and major diseases.
RNA level application
Transcriptome sequencing
Transcriptome sequencing, also known as RNA-seq or mRNA-seq, namely enrich single-stranded mRNA from total RNA, then reverse transcription into doublestranded cDNA, then which will be used to highthroughput sequencing and subsequent correlation analysis. Transcriptome is the foundation and starting point for studying gene function and structure. With reference genome squence, scientists can obtain much more information, such as gene expression, alternative splicing, optimizing-gene structure, and new genes by comparing RNA-seq data with genomic DNA sequences. For no reference genome species, de novo sequencing would play an important role in transcriptome studies, and would be effectively used to discover new genes and develop new molecular markers. Guo et al. (2011) performed half Roche/454 GS-FLX to identify more than 5 000 Simple Sequence Repeats (SSRs). Transcriptional regulation is the most important regulation, and transcriptome sequencing studies built on the basis of high-throughput sequencing have gradually substituted for gene chip technology to be one of the current main approaches to study gene expression on the level of the whole-genome. By transcriptome sequencing, researchers could getabundance expression of transcript, transcriptional loci, alternative splicing, SNP and other important information. Zhang et al. (2010) took Oryza sativa L. ssp. indica cv. 9311 as material to researching rice transcriptome , they extracted total RNA from callus, root at seedling stage of 14 days, shoot at seedling stage of 14 days, flag leaves at tillering stage, flag leaves at flowering stage, panicle at booting stage, panicle at flowering stage, and panicle at filling stage, then sequenced the total RNA from each sample and showed transcriptome map of different organs of cultivated rice. They detected 7 232 novel transcript units, which have low abundance of expression and tissue specificity, 23 800 alternative splicing occurred in 33% of rice genome, 1 356 highly reliable chimeric fusions, and 234 candidate chimeric transcripts, suggesting that the transcriptional fusion was more common than expected to occur, those data provided a stable foundation for future functional studies upon complex mechanisms of transcriptional regulation of rice. Besides that, the technology has been applied in potato (Shan et al., 2013), watermelon (Grassi et al., 2013; Guo et al., 2011), pea (Liu et al., 2013), green tea (Pan et al., 2014) and so on.
Digital gene expression profiling technology
Digital Gene Expression (DGE) is to construct nonbias cDNA library of the cells or tissue in a particular state, through large-scale cDNA sequencing, collection of cDNA sequence fragments, and the qualitative and quantitative analysis about its mRNA population, scientists could obtain types of gene expression and abundance information of the certain cell or tissue in the state. Gene expression profiling in the past mainly relied on conventional microarray technology, which relied on known gene sequences to design probes with fluorescence labeling and hybridization, then calculated the amount of expression according to fluorescence intensity, whereas its error was huge, and it has no ability to detect unknown gene expression. In contrast, DGE is more sensitive, accurate and suitable for comparing gene expression studies, and gradually takes the place of the gene chip technology in order to avoid many of the inherent limitations of microarray analyses. The combination of transcriptome sequencing and DGE technologies can effectively explore new functional genes for species with reference genome or without. Luan et al. (2011) used DGE method to analyze the gene expression variations between the nonviruliferous and viruliferous whiteflie, then they revealed the relationship of coevolved adaptations between begomoviruses and whiteflies and would provide a road map for future investigations into the complex interactions between plant viruses and their insect vectors. In addition, Gao et al. (2014) performed DGE to investigate the gene expression profiles of 4008 and p50 silkworm strains to provide important clues on the molecular mechanism of BmCPV invasion and resistance mechanism of silkworms against BmCPV infection. Yan et al. (2014) applied comparative DGE and quantitative real time PCR to figure out five transcripts encoding proteins putatively associated with scent biosynthesis in roses and provided a foundation for scent-related gene discovery in roses.
MicroRNA sequencing
MicroRNA is a non-coding RNA, only about 20-30 nucleotides, while it plays a significant role in vivo. Post-transcriptional gene regulation by microRNA is a novel biological mechanism of gene regulation. In recent years, the technology has been brought into a wide focus in the scientific community. And the rise of high-throughput sequencing has brought new ideas to microRNA research. Although the high-throughput sequencing has the bottleneck of short sequence difficult to break, it is very suitable for sequencing microRNA. Researchers could take its advantage to predict new microRNA, research conserved microRNA, establish microRNA expression profiling, compare miRNA expression abundance as well as find other non-coding RNA through sequencing. Wei et al. (2009) used high-throughput sequencing to research small RNAs of the locusts. By similarity searching against microRBase database, they identified 50 conserved microRNA families, and identified 185unique microRNAs families of locust through bioinformatics analysis. And the analysis of microRNAs expression between gregarious and solitary locust revealed that microRNAs expression of the solitary is richer than the gregarious, and drew expression profiles of microRNAs of two different lifestyles. The technology, currently, has been succeeded in researching rice (Hu et al., 2014; Liu et al., 2014; Mittal et al., 2013; You et al., 2014) and peach (Zhenlin, 2013).
Epigenomics applications
Chromatin immunoprecipitation sequencing
Chromatin immunoprecipitation sequencing (ChIPSeq) technology is a powerful tool to study the interactions between protein and DNA in vivo, which combines the advantages of both Chromatin immunoprecipitation (ChIP) and high-throughput sequencing technology and have been successfully applied in the genome-wide study, such as protein binding sites, transcription factor binding sites and specific histone modification sites studies. Thus, scientists could get the information from segment of DNA interacted with transcription factors or histone in full genome-wide. Li et al. (2014) used ChIP-seq to predict estrogen receptor (ER) biding sites in human breast cancer cell line MCF7, their result showed that E2 stimulated breast cancer cell growth through ER, which might infer the function of ER in occurrence and development of breast cancer. In recent years, ChIP-Seq has also been applied mainly in studies on rats (Corbo et al., 2010; Hull et al., 2013; Rintisch et al., 2014; Triff et al., 2013) and human (Liu and Cheung, 2014; Pinho et al., 2013; Xing et al., 2013; Zheng et al., 2013).
DNA methylation sequencing
DNA methylation is another important way of gene regulation, which can control gene expression by altering chromatin structure, stability of DNA and DNA-protein interactions. Currently, there are at least three kinds of DNA methylation analysis technique established on high-throughput sequencing: methylated DNA immunoprecipitation sequencing (MeDIP-Seq) (Down et al., 2008), methyl-binding protein sequencing (MBD-Seq) and bisulfite sequencing (BS-Seq) (Cokus et al., 2008). Highthroughput sequencing technology also provides an efficient solution to detect genome-wide methylation sites. Taylor et al. (2007) applied 454 sequencing technology to reveal an association between a single nucleotide polymorphism and the methylation present in LRP1B promoter. They finally concluded that this new generation of methylome sequencing would provide digital profiles of the aberrant DNA methylation for individual human cancers and offer a robust method for the epigenetic classification of tumor subtypes. Currently, DNA methylation sequencing technology has achieved fruitful research results in DNA methylation studies on CpG (Li et al., 2013; Nan et al., 1998; Shanmuganathan et al., 2013), cancer (Calcagno et al., 2013), and provided an important alternative to conventional approaches in human brain studies (Houston et al., 2013).
Conclusions
High-throughput sequencing technology is still at its early stage of development, but we could foresee that it will be the golden time for the rapid development of the third generation sequencing technology and the coexistence of sequencing technologies of three generations in the next few years. With the appearance of new sequencing technologies, the cost of sequencing would continue to decline rapidly. Development of new drug for incurable diseases, molecular breeding technology for fine breeds will become easier and faster. Therefore, scientists in different fields will be allowed to spend less and less money on sequencing genome or transcriptome of species to achieve better experimental design and obtain more new discoveries. In addition, how to analyze the massive sequencing data generated by high-throughput sequencing technology and extract valuable bio-information from will become a hot research in the future.
References
Aksyonov S A, Bittner M, Bloom L B, et al. 2006. Multiplexed DNA sequencing-by-synthesis. Analytical Biochemistry, 348(1): 127-138.
Altermann E and Klaenhammer T R. 2005. Pathway voyager: pathway mapping using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. BMC Genomics, 6: 49-55.
Ansorge W J. 2009. Next-generation DNA sequencing techniques. New Biotechnology, 25(4): 195-203.
Ashburner M, Ball C A, Blake J A, et al. 2000. Gene ontology: tool for the unification of biology. Nature genetics, 25(1): 25-29.
Birol I, Jackman S D, Nielsen C B, et al. 2009. De novo transcriptome assembly with ABySS. Bioinformatics, 25(21): 2872-2877.
Butler J, MacCallum I, Kleber M, et al. 2008. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res, 18(5): 810-820.
Calcagno D Q, Gigek C O, Chen E S, et al. 2013. DNA and histone methylation in gastric carcinogenesis. World Journal of Gastroenterology, 19(8): 1182-1192.
Cheung F, Haas B J, Goldberg S M D, et al. 2006. Sequencing medicago truncatula expressed sequenced tags using 454 life sciences technology. BMC Genomics, 7: 272-283.
Cokus S J, Feng S, Zhang X, et al. 2008. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature, 452(7184): 215-219.
Conesa A, Gotz S, Garcia-Gomez J M, et al. 2005. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics, 21(18): 3674-3676.
Corbo J C, Lawrence K A, Karlstetter M, et al. 2010. ChIP-seq reveals the cis-regulatory architecture of mouse photoreceptors. Genome Research, 20(11): 1512-1525.
Dennis G, Sherman B T, Hosack D A, et al. 2003. DAVID: database for annotation, visualization, and integrated discovery. Genome Biology, 4(9): 12-22.
Down T A, Rakyan V K, Turner D J, et al. 2008. A bayesian deconvolution strategy for immunoprecipitation-based DNA methylome analysis. Nature Biotechnology, 26(7): 779-785.
Eid J, Fehr A, Gray J, et al. 2009. Real-time DNA sequencing from single polymerase molecules. Science, 323(5910): 133-138.
Gao K, Deng X, Qian H, et al. 2014. Cytoplasmic polyhedrosis virusinduced differential gene expression in two silkworm strains of different susceptibilities. Gene, 539(2): 230-237.
Grabherr M G, Haas B J, Yassour M, et al. 2011. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nature Biotechnology, 29(7): 644-650.
Grassi S, Piro G, Lee J M, et al. 2013. Comparative genomics reveals candidate carotenoid pathway regulators of ripening watermelon fruit. BMC Genomics, 14(1): 781-793.
Guo S, Liu J, Zheng Y, et al. 2011. Characterization of transcriptome dynamics during watermelon fruit development: sequencing, assembly, annotation and gene expression profiles. BMC Genomics, 12: 454.
Guo S, Zhang J, Sun H, et al. 2013. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat Genet, 45(1): 51-58.
Harris T D, Buzby P R, Babcock H, et al. 2008. Single-molecule DNA sequencing of a viral genome. Science, 320(5872): 106-109.
Houston I, Peter C J, Mitchell A, et al. 2013. Epigenetics in the human brain. Neuropsychopharmacology, 38(1): 183-197.
Hu W, Wang T, Yue E, et al. 2014. Flexible microRNA arm selection in rice. Biochemical and Biophysical Research Communications, 447(3): 526-530.
Hu Z, Hung J-H, Wang Y, et al. 2009. VisANT 3.5: multi-scale network visualization, analysis and inference based on the gene ontology. Nucleic Acids Research, 37: 115-121.
Huang S, Li R, Zhang Z, et al. 2009. The genome of the cucumber, Cucumis sativus L. Nat Genet, 41(12): 1275-1281.
Hull R P, Srivastava P K, Souza Z, et al. 2013. Combined ChIP-seq and transcriptome analysis identifies AP-1/JunD as a primary regulator of oxidative stress and IL-1 beta synthesis in macrophages. BMC Genomics, 14: 5-16.
Iseli C, Jongeneel C V, Bucher P. 1999. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proceedings International Conference on Intelligent Systems for Molecular Biology; ISMB. International Conference on Intelligent Systems for Molecular Biology, 12: 138-148.
Kahvejian A, Quackenbush J, Thompson J F. 2008. What would you do if you could sequence everything. Nature Biotechnology, 26(10): 1125-1133.
Langmead B, Trapnell C, Pop M, et al. 2009. Ultrafast and memoryefficient alignment of short DNA sequences to the human genome. Genome Biol, 10(3): 25-29.
Li H, Durbin R. 2009. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics, 25(14): 1754-1760.
Li H, Ruan J and Durbin R. 2008. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Research, 18(11): 1851-1858.
Li Q, Wang H, Yu L, et al. 2014. ChIP-seq predicted estrogen receptor biding sites in human breast cancer cell line MCF7. Tumor Biology, 35(5): 4779-4784.
Li R, Yu C, Li Y, et al. 2009. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics, 25(15): 1966-1967.
Li Z-G, Jiao Y, Li W-J, et al. 2013. Hypermethylation of two CpG sites upstream of CASP8AP2 promoter influences gene expression and treatment outcome in childhood acute lymphoblastic leukemia. Leukemia Research, 37(10): 1287-1293.
Lin H, Zhang Z, Zhang M Q, et al. 2008. ZOOM! Zillions of oligos mapped. Bioinformatics, 24(21): 2431-2437.
Liu H, Guo S, Xu Y, et al. 2014. OsmiR396d regulated OsGRFs function in floral organogenesis in rice through binding to their targets OsJMJ706 and OsCR4. Plant Physiology, 165(1): 160-174.
Liu M H, Cheung E. 2014. Estrogen receptor-mediated long-range chromatin interactions and transcription in breast cancer. Molecular and Cellular Endocrinology, 382(1): 624-632.
Liu Z, Ma L, Nan Z, et al. 2013. Comparative transcriptional profiling provides insights into the evolution and development of the zygomorphic flower of Vicia sativa (Papilionoideae). PLoS One, 8(2): 573-588.
Luan J-B, Li J-M, Varela N, et al. 2011. Global analysis of the transcriptional response of whitefly to tomato yellow leaf curl china virus reveals the relationship of coevolved adaptations. Journal of Virology, 85(7): 3330-3340.
Maxam A M and Gilbert W. 1977. A new method for sequencing DNA. Proceedings of the National Academy of Sciences of the United States of America, 74(2): 560-564.
Mittal D, Mukherjee S K, Vasudevan M, et al. 2013. Identification of tissue-preferential expression patterns of rice miRNAs. Journal of Cellular Biochemistry, 114(9): 2071-2081.
Nan X, Ng H H, Johnson C A, et al. 1998. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature, 393(6683): 386-389.
Nirenberg M, Caskey T, Marshall R, et al. 1966. The RNA code and protein synthesis. Cold Spring Harbor Symposia on Quantitative Biology, 31: 11-24.
Pan J, Zhang Q, Xiong D, et al. 2014. Transcriptomic analysis by RNA-seq reveals AP-1 pathway as key regulator that green tea may rely on to inhibit lung tumorigenesis. Molecular Carcinogenesis, 53(1): 19-29.
Pinho F G, Frampton A E, Nunes J, et al. 2013. Downregulation of microRNA-515-5p by the estrogen receptor modulates sphingosine kinase 1 and breast cancer cell proliferation. Cancer Research, 73(19): 5936-5948.
Rasmussen M, Li Y, Lindgreen S, et al. 2010. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature, 463(7282): 757-762.
Rintisch C, Heinig M, Bauerfeind A, et al. 2014. Natural variation of histone modification and its impact on gene expression in the rat genome. Genome Research, 24(6): 942-953.
Rusk N. 2009. Cheap third-generation sequencing. Nature Methods, 6(4): 244-245.
Sanger F. 1988. Sequences, sequences, and sequences. Science, 280(5369): 1515-1515.
Sanger F, Nicklen S and Coulson A R. 1977. DNA sequencing with chain-terminating inhibitors. Proceedings of the National Academy of Sciences of the United States of America, 74(12): 5463-5467.
Schuster S C. 2008. Next-generation sequencing transforms today's biology. Nature Methods, 5(1): 16-18.
Shan J, Song W, Zhou J, et al. 2013. Transcriptome analysis reveals novel genes potentially involved in photoperiodic tuberization in potato. Genomics, 102(4): 388-396.
Shanmuganathan R, Basheer N B, Amirthalingam L, et al. 2013. Conventional and nanotechniques for DNA methylation profiling. Journal of Molecular Diagnostics, 15(1): 17-26.
Sultan M, Schulz M H, Richard H, et al. 2008. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science, 321(5891): 956-960.
Taylor K H, Kramer R S, Davis J W, et al. 2007. Ultradeep bisulfite sequencing analysis of DNA methylation patterns in multiple gene promoters by 454 sequencing. Cancer Res, 67(18): 8511-8518.
Trapnell C, Roberts A, Goff L, et al. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nature Protocols, 7(3): 562-578.
Triff K, Konganti K, Gaddis S, et al. 2013. Genome-wide analysis of the rat colon reveals proximal-distal differences in histone modifications and proto-oncogene expression. Physiological Genomics, 45(24): 1229-1243.
Velasco R, Zharkikh A, Affourtit J, et al. 2010. The genome of the domesticated apple (Malus x domestica Borkh). Nature Genetics, 42(10): 833-840.
Wang X, Wang H, Wang J, et al. 2011. The genome of the mesopolyploid crop species Brassica rapa. Nature Genetics, 43(10): 1035-1157.
Watson J D and Crick F H. 1953. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature, 171(4356): 737-738.
Weber A P M, Weber K L, Carr K, et al. 2007. Sampling the arabidopsis transcriptome with massively parallel pyrosequencing. Plant Physiology, 144(1): 32-42.
Wei Y, Chen S, Yang P, et al. 2009. Characterization and comparative profiling of the small RNA transcriptomes in two phases of locust. Genome Biology, 10(1): 45-60.
Wheeler D A, Srinivasan M, Egholm M, et al. 2008. The complete genome of an individual by massively parallel DNA sequencing. Nature, 452(7189): 872-885.
Xing Y, Yang Y, Zhou F, et al. 2013. Characterization of genome-wide binding of NF-kappa B in TNF alpha-stimulated HeLa cells. Gene, 526(2): 142-149.
Xu X, Pan S, Cheng S, et al. 2011. Genome sequence and analysis of the tuber crop potato. Nature, 475(7355): 189-194.
Yan H, Zhang H, Chen M, et al. 2014. Transcriptome and gene expression analysis during flower blooming in Rosa chinensis 'Pallida'. Gene, 540(1): 96-103.
Ye J, Fang L, Zheng H, et al. 2006. WEGO: a web tool for plotting GO annotations. Nucleic Acids Research, 34: 293-297.
You J, Zong W, Du H, et al. 2014. A special member of the rice SRO family, OsSRO1c, mediates responses to multiple abiotic stresses through interaction with various transcription factors. Plant Molecular Biology, 84(6): 693-705.
Zeeberg B R, Feng W, Wang G, et al. 2003. GoMiner: a resource forbiological interpretation of genomic and proteomic data. Genome Biol, 4(2): 28-32.
Zhang G, Guo G, Hu X, et al. 2010. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Geome Res, 20(5): 646-654.
Zheng L Y, Guo X S, He B, et al. 2011. Gemome-wide patterns of genetic variation in sweet and grain sorghum (Sorghum bicolor). Geome Biol, 12(11): 114-120.
Zheng Y, Zha Y, Spaapen R M, et al. 2013. Egr2-dependent gene expression profiling and ChIP-Seq reveal novel biologic targets in T cell anergy. Molecular Immunology, 56(4): 530-536.
Zhenlin W. 2013. Identification and characterization of microRNAs and their targets in peach (Prunus persica). International Journal of Agriculture and Biology, 15(5): 1017-1020.
Zhou X G, Ren L F, Li Y T, et al. 2010. The next-generation sequencing technology: a technology review and future perspective. Sci China Life Sci, 53(1): 13-25.

Fig. 2 Observation of mitochondria by Janus Green B reactive dying
Wang Shuai et al. Method for Isolating Mitochondrial DNA from Etiolated Tissue of Cabbage. 2014, 21(3): 23-29.

Fig. 4 12 h post FUW-EGFP and FUW-tetO-EGFP lentivirus infection (DOX induction), GFP specifically expressed in TE
Fig. 5 TE specific functional gene manipulation
Yin Zhi et al. Lentivirus Mediated Gene Manipulation in Trophectoderm of Porcine Embryos. 2014, 21(3): 39-45.
S6
A
1006-8104(2014)-03-0084-13
Received 29 October 2013
Supported by the National Natural Science Foundations of China (31272186; 31301791)
Zhu Qiang-long (1989-), male, Master, engaged in the research of bioinformatics and watermelon molecular breeding. E-mail: longzhu2011@126.com
* Corresponding author. Luan Fei-shi, professor, supervisor of Ph. D students, engaged in the research of watermelon and melon molecular breeding. E-mail: luanfeishi@sina.com
杂志排行

Journal of Northeast Agricultural University(English Edition)的其它文章
- Effect of Seed Soaking with Exogenous Proline on Seed Germination of Rice Under Salt Stress
- Physiological Changes and Cold Tolerance of Three Camphor Species During Natural Winter Temperature Fluctuations
- Effects of Methylated Soybean Oil Adjuvant on Fomesafen Efficacy to Weeds
- Method for Isolating Mitochondrial DNA from Etiolated Tissue of Cabbage
- Lentivirus Mediated Gene Manipulation in Trophectoderm of Porcine Embryos
- Effects of Allicin on Lipid Metabolism and Antioxidant Activity in Chickens