PP瓶包装输液产品灭菌工艺的可靠性探讨
2014-03-06马晓红王忠宝侯仁秀
马晓红 王忠宝 张 刚 侯仁秀
(山东齐都药业有限公司,山东淄博 255400)
0 引言
大容积水浴式灭菌器是目前国际上对输液产品进行最后灭菌处理的最先进的设备,我公司生产PP瓶包装输液产品,根据其生产工艺及无菌药品质量要求,采用大容积水浴式灭菌器对产品进行最终灭菌处理。灭菌工艺的目的是使具有一定微生物污染指数的产品,经过灭菌处理后,既能达到无菌保证水平要求的10-6,又能保证产品或PP瓶不因受热而发生降解,确保用药安全性。基于风险的角度,本文以PP瓶包装输液大容积水浴式灭菌器为模型,对影响灭菌工艺可靠性的风险因素进行分析,并有针对性地制定风险控制措施,将影响最终灭菌效果的风险降到最低。
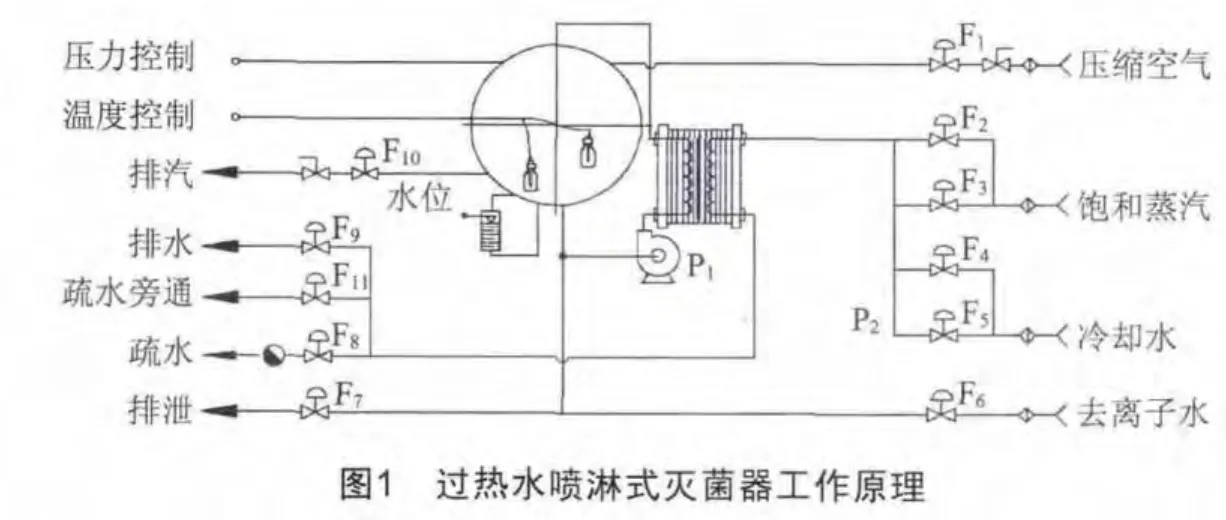
1 PP瓶输液大容积水浴式灭菌器的构造及工作原理
大容积水浴式灭菌器由硬件系统和控制系统构成,过热水喷淋式灭菌器的工作原理如图1所示。
灭菌器的硬件部分包括主体、密封门、水泵、换热器、灭菌车和管路系统,控制系统由现场控制系统(PLC)、上位机和记录仪组成。采用高温过热水喷淋方式对输液产品进行加热和灭菌,特点是温度分布均匀、温度控制范围广,与蒸汽、冷却水完全隔离,污染少,是一种高性能、高智能化的灭菌设备。由于灭菌过程中采用独特的压力平衡技术,从而保证了产品灭菌后仍能保持良好的形状,是塑瓶产品的首选灭菌方法。
2 风险因素分析
灭菌工艺可靠性风险因素分析如图2所示。
通过因素分析图可以看出,灭菌设备的适用性、可验证性、验证状态维护、工艺参数的科学合理性、监测系统的精度、人员操作的规范性和待灭菌产品的微生物污染指数及热稳定性,都是影响灭菌工艺可靠性的主要因素。
2.1 风险控制
2.1.1 设备适用性是灭菌工艺可靠性的硬件保障
灭菌设备的适用性指灭菌器是执行灭菌工艺的能力,即灭菌设备的灭菌工艺各参数的控制准确度和精确度能否使任意位置的产品的实际F0值符合灭菌工艺规定的F0值范围。基于“质量源于设计”与“风险管理”的理念,充分理解工艺及设备验证,设备供应商应根据URS(用户需求)提供FAT(工厂现场测试)、SAT(终端用户测试),且FAT和SAT的全部数据和测试报告应由终端用户的质量部门批准[1]。关键。灭菌器作为PP塑瓶输液产品生产过程中的关键设备,在进行了充分的灭菌器性能验证、产品工艺验证后,应按照验证所确定的工艺参数、装载方式、装载量、取样点等制定实用的SOP,操作人员经培训具备相适应的操作能力,以便设备的日常运行保持在验证范围内。验证状态的维护关键在于日常操作规范化、设备校准实效化。

日常操作规范化,即进行正确的操作和必要的巡回检查,班前班后对设备进行必要的卫生清理和清洁,每班对设备的易松动部件进行检查和紧固,定期对设备进行预防性维护。设备校准实效化,即按照规定对设备的仪器仪表进行校验,对设备的温度控制系统进行定期校准,对设备的连锁保护系统进行定期核查。同时根据GMP要求,每年对大容积水浴灭菌器进行再验证,确保其性能满足工艺要求。当设备发生重大变更或相应的装载方式、装载量发生变更时均需进行验证,以保证灭菌工艺的可靠性,从而达到规定的灭菌效果。
2.1.2 确定灭菌工艺参数并进行监测和监控是确保灭菌工艺可靠执行的系统保障
根据待灭菌产品的特性,确定科学合理的灭菌工艺参数,并能准确有效地进行监测和监控,是确保灭菌工艺可靠执行的系统保障。
2.1.2.1 注射剂的不同分类
注射剂按照药物制剂灭菌工艺的不同分为最终灭菌产品和非最终灭菌产品。我公司生产的PP塑瓶输液产品,根据其生产工艺流程及无菌药品质量要求,遵循“质量源于设计”的风险管理理念,按照“湿热灭菌程序决策树”[3](图3)的原则,综合考虑PP塑瓶物理性质的影响(温度、压力的不当会导致容器变形),确定PP塑瓶输液产品灭菌工艺采用过热水湿热灭菌程序,灭菌工艺参数确定为115℃、30/40 m in、F0≥8的残存概率法进行灭菌程序的控制与实施。
2.1.1.1 制造工厂现场测试
FAT即制造工厂现场测试,是设计确认的开始,是确认设备制造商是否按照已经过用户确认的设计标准进行加工制造,应当由灭菌器供应商独立完成,针对每个规格型号(或容积)的灭菌器通过较密集设置的温度传感器进行灭菌器内温度分布研究,建议每立方米不少于1个传感器,找出代表性的温度点,即最快到达灭菌温度的点、最慢到达灭菌温度的点、降温时最慢到达90℃的点、灭菌阶段最冷点、灭菌阶段最热点等[2],并通过多次运行证明其重现性,确定灭菌器控制传感器的位置和数量。测试可以分次或分模块进行,但每次测试应至少有2个传感器放置在上一次测试的相同位置(其中1个放置在排水口),以证明重现性和分次试验数据的相关性,还应当包含空载热分布研究,给出满载热分布和热穿透验证时放置验证传感器的位置。
2.1.1.2 终端用户测试
SAT即终端用户测试,是在FAT测试结果符合设计标准后,设备运达用户现场,并与用户共同对公用系统能力进行确认,完成安装后进行的测试。灭菌器供应商明确灭菌器内部及外部所有传感器的数量和位置,并提供安装位置的安装依据和确认证明文件,根据双方约定进行空载或满载热分布研究,提供水均匀的相关测试和证明文件,如柜内各个喷淋口、喷淋盘布水、灭菌车多层结构布水等均匀性证明,为验证打好基础。
2.1.1.3 验证及验证状态维护

验证及验证状态维护,是确保灭菌工艺可靠性的
2.1.2.2 灭菌工艺的无菌保证与产品稳定性
为充分保证待灭菌产品的无菌保证水平与灭菌后产品的稳定性,在进行产品灭菌工艺验证时要充分考虑3批产品的热穿透与生物挑战性实验。
热穿透验证项目应包括每种装载容器(500 m L、250m L、100m L)的装载方式、每种方式的运行次数、每种方式的冷点是否确定等,以及出现热穿透偏差如何处理。若在同一灭菌柜进行多品种多规格的灭菌再验证时应根据风险分析的方法,筛选出最有效、最经济的验证方案[3]。温度测量系统(灭菌器的温度传感器和验证仪Pt铂电阻)应提供每一测点的温度数据并进行数据分析,确定相对冷点对产品灭菌效果的影响及日常无菌取样的位置,便于验证后的无菌效果监测,同时确定相对热点对产品性质的影响,避免造成产品的降解等反应。经过对热穿透的总体评价确定最佳灭菌效果的装载量,以指导今后的生产。
生物挑战实验:灭菌工艺与灭菌前微生物的控制也有较大的关联,众所周知,气源性微生物革兰氏阳性菌较多,它们可形成芽孢,水源性革兰氏则阴性菌居多,不会形成芽孢,耐热性差,但会产生细菌内毒素。影响芽孢耐热性的因素主要是芽孢存在的介质、受热时的湿度。当高温破坏了细胞中起生命作用的蛋白质、酶及核酸时,细胞就会死亡。湿热灭菌的原理也是基于此。
由于采取过度杀灭法(121℃、12m in)的灭菌程序能够保证较高的无菌保证值,因此对被灭菌品不需要进行常规的初始菌监控。而残留概率法由于其F0值<8,就需要对被灭菌物品的微生物数量和耐热菌进行控制。因此,塑瓶生产过程虽然经过配制过滤系统的除菌过滤,微生物初始数量较低,但是由于其灭菌工艺参数不是过热灭菌的参数,因此确定对每批产品灌装开始、中间、结束进行微生物限度的监测,并在进行产品灭菌工艺验证时进行耐热菌的检查,通过监控初始微生物污染水平与检出菌的耐热性保证产品的无菌水平。
GMP要求在进行产品灭菌工艺验证时必须使用生物指示剂监控灭菌工艺,因此必须做好生物指示剂的筛选与放置位置的评估确定[3]。通常根据产品的耐热性及所筛选的灭菌参数,选择与产品灭菌工艺参数相匹配的生物指示剂与产品一起灭菌,当生物指示剂培养结果为阴性时,结合灭菌前初始菌的微生物负荷和是否存在耐热菌和生物指示剂的结果,为产品的无菌提供依据。
生物指示剂的选型与放置位置对验证结果的准确性至关重要,湿热灭菌耐热性强的孢子主要有2个属:梭菌属,它一般是严格的厌氧菌(anaerobe);芽孢杆菌属,是需氧菌(aerobe),它可能是兼性厌氧菌。最常用的菌是枯草芽孢杆菌、嗜热脂肪芽孢杆菌,PP塑瓶产品根据其产品特性通常使用嗜热脂肪芽孢杆菌作为生物指示剂。其放置位置应根据被灭菌产品的情况而定,除了在灭菌器腔室内均匀分布外,还要考虑放置最难灭菌部位及冷点位置。同时应以冷点温度作为程序控制的依据,当所有测试点均达到灭菌温度时才计算标准灭菌时间。任何新的工艺都需验证其有效性。
2.1.2.3 自控系统的运行
灭菌工艺的可靠性是通过自控系统的稳定运行来实现的,而灭菌程序控制与记录仪表、温度传感器的校验是监测灭菌工艺可靠性的尺度。灭菌过程中,要使用适当精密度和准确度的设备对关键运行参数进行记录,特别是在被灭菌品或灭菌腔室冷点处安装温度探头,通过记录仪记录该点的温度情况,该冷点通过验证确定。为提高此温度参数的可靠性,应在同一部位安放另一个独立的测温探头,用显示仪表显示温度数值,供操作人员直接与记录仪记录的趋势图进行对照。温控仪趋势图应与灭菌图谱同时纳入批生产记录中[4]。另外灭菌柜温度传感器的校准应每半年校验1次,确保其准确度。特别是在使用过程中其导线部分,其材质必须耐温、耐压和耐湿。在使用过程中当导线软管部分逐步老化时,其性能下降,水蒸气就可能进入软管内部,因此会导致电阻值漂移,干扰测量、控制系统的稳定性与准确性,因此要选用质量较好的测温系统(包括控制、记录)。测温系统(通过变送器调节或在程序中进行补偿)的校准需要由企业技术人员或更高等级并能溯源至国家标准的恒温浴、干井等仪器完成。如果校准时发现灭菌器测温系统的测量误差超出最大允许误差范围,应进行风险评估,对之前灭菌的产品及运行参数进行追溯,以保证其不会危及产品的无菌性和稳定性。
2.1.3 设备是基础,人员是关键
灭菌程序的可靠性源于自控系统的程序设计与运行过程的可靠,应综合考虑温控系统、动力系统、权限控制、动力故障、警告和报警、断电保护等项目,全面评估后将其纳入URS的要求中,在设计阶段充分考虑风险及潜在风险,避免人员的误操作或故障造成风险。为了降低人员带来的风险,应当着力抓好人员资质管理与培训,确保灭菌岗位操作人员经过必要的培训考核合格后持证上岗。
灭菌程序要依靠灭菌设备的有效运行、温控探头的准确性来实施,因此所有涉及灭菌程序实施的蒸汽压力、压缩空气压力、水源、上位机控制系统、温控系统、温度传感器等均需进行启动前的确认,保证整个过程的实施符合工艺需求,因此设备的验证、日常维护保养、巡检应严格按照SOP执行,人员的操作、复核、升温阶段、灭菌阶段、冷却阶段的观察均严格按照SOP执行,不得私自采取手动操作,影响灭菌效果。工作人员应加强培训与学习,微机操作人员要善于积累工作经验,提高预防异常情况发生的能力。SOP的修订要不断地更新,使其更适应灭菌工艺的执行,人员的培训应定期进行,确保执行灭菌工艺无偏移。
基于风险的理念,应定期对塑瓶输液产品的焊盖工艺的参数进行验证,确保塑瓶的密封完整性,保证其在药品有效期内有完好的密封性,防止灭菌过程、贮存过程微生物的侵入,可按照“药品生产验证指南”[5]的要求进行微生物挑战实验,确定其密封完整性。若焊盖工艺发生变更时,必须评估其对密封完好性的影响,必要时对该项进行验证。另外,为了防止产品经灭菌后受到二次污染的风险,应定期清洁、维护灭菌设备,检测灭菌循环水的微生物限度,确保接触被灭菌品的水质未发生微生物污染。
3 结语
综上所述,PP塑瓶输液产品灭菌程序的可靠性保证应重点做好灭菌设备的URS、FAT、SAT测试,做好灭菌设备验证工作及温度传感器的校准,保持其验证状态,并根据产品特性进行满载热穿透实验和生物指示剂验证,确定产品灭菌工艺的可靠性。通过设备的稳定运行、人员的规范操作,严格执行灭菌工艺。定期对灭菌工艺、待灭菌产品的密封性、灭菌循环水的微生物负荷进行检测,在保证灭菌程序可靠运行的基础上,更好地保证PP塑瓶产品的稳定性。
[1]国家食品药品监督管理局药品认证管理中心.无菌药品[M].北京:中国医药科技出版社,2011
[2]中国医药设备工程协会.水浴式灭菌柜选型用户需求(试行)[S]
[3]国家食品药品监督管理局.药品生产质量管理规范(2010年修订)[S]
[4]中国医药设备工程协会.湿热灭菌工艺验证指南[S]
[5]国家食品药品监督管理局药品认证管理中心.药品生产验证指南[M].北京:化学工业出版社,2003
