cry6Aa2m基因植物表达载体构建及大豆的遗传转化
2014-01-14纪巍杨丽坤柏锡朱延明才华
纪巍,杨丽坤,柏锡,朱延明,才华
(东北农业大学生命科学学院,哈尔滨 150030)
cry6Aa2m基因植物表达载体构建及大豆的遗传转化
纪巍,杨丽坤,柏锡,朱延明,才华
(东北农业大学生命科学学院,哈尔滨 150030)
cry6Aa2杀线虫晶体蛋白基因对线虫有很高的杀虫活性,根据大豆密码子的偏爱性,人工合成杀线虫最小毒力区间cry6Aa2m基因。以pCAMBIA3300为骨架,构建由组成型强启动子E12 CaMV35S调控的cry6Aa2m基因、筛选标记基因为除草剂抗性bar基因的植物表达载体pCBE-cry6,用农杆菌介导法对东农50大豆子叶节进行遗传转化,通过PCR和RT-PCR检测,结果证明cry6Aa2m基因已整合到大豆基因组中并进行转录,获得转cry6Aa2m基因大豆新株系18株,为大豆抗线虫分子育种研究提供材料。
大豆;cry6Aa2m基因;遗传转化;农杆菌介导;转基因株系
大豆胞囊线虫(Heterodera glycines)是一种大豆毁灭性病害,线虫寄生后对根部造成机械损伤,严重阻碍水分和无机盐从根部吸收和向上运输,同时产生刺激植物的物质,引起寄主植物畸形,如虫瘿、根结、胞囊,影响大豆产量,并引发其他病害并发感染[1]。大豆胞囊线虫病对我国大豆产量和品质构成严重威胁。
苏云金芽胞杆菌作为目前世界上应用最广泛、最成功的生物杀虫剂和转基因抗虫育种原动力资源库,保持近90年的安全高效使用记录[2],对多种昆虫、线虫、螨类和原生动物等具有特异性杀虫活性[3-4]。目前发掘的杀线虫基因还很少,其中cry1、cry5、cry6、cry12、cry13、cry14、cry21是现有具有杀线虫能力的晶体蛋白基因,cry6与其他cry类基因亲缘关系较远,蛋白质结构上与其他杀虫晶体蛋白差异较大,单独作为cry6亚家族;其余被归为cry5亚家族[5]。Li等把cry6A基因基于植物密码子偏好性进行密码子优化,由强组成型启动子35S启动并且转化番茄,结果显示cry6A基因在番茄根中大量表达,而且线虫能摄取54 ku的cry6A晶体蛋白,与对照相比线虫产卵数减少4倍[6]。Li等把cry5B成功转化番茄,晶体蛋白表达后番茄根中的线虫产卵数降低3倍[7]。表明晶体蛋白在转基因植物中具有杀线虫的能力。
cry6Aa2基因是从对北方根结线虫有高毒力的苏云金芽胞杆菌野生型菌株YBT-1518中最新克隆到的一个Bt基因[8],通过杀虫活性测定表明,晶体蛋白cry6Aa2对线虫有高毒力杀虫能力,半致死浓度为23.9 μg·mL-1[9]。本研究根据大豆密码子偏爱性,人工合成cry6Aa2基因的最小毒力区间,利用东北农业大学植物基因工程与分子生物学研究室已建立的高效大豆遗传转化体系(农杆菌介导法),转化大豆东农50,通过PCR技术筛选和验证,初步获得转基因大豆新株系,对大豆胞囊线虫的防治、大豆产量和品质提高,具有重要的研究价值和广阔的应用开发前景。
1 材料与方法
1.1 材料
1.1.1 植物材料
大豆品种东农50,由东北农业大学大豆研究所提供。
1.1.2 培养基
所用培养基及其成分参见文献[10-11]方法。
1.1.3 菌株和质粒
大肠杆菌DH5α,农杆菌EHA105由东北农业大学植物生物工程研究室保存。cry6Aa2基因由华中农业大学农业微生物学国家重点实验室提供。以pCAMBIA3300为骨架,构建由强组成型启动子E12 CaMV35S调控的含有cry6Aa2m目的基因、除草剂抗性Bar基因为筛选标记基因的植物表达载体pCBE-cry6,参考Hofgen和Willmitzer冻融法[12]直接将质粒导入农杆菌菌株中,其T-DNA区的结构如图1所示。

图1 质粒pCBE-cry6的T-DNA区Fig.1 Linear map of plant expression plasmid pCBE-cry6 T-DNA cassette
1.2 试验方法
1.2.1 密码子改造
基于大豆基因组中最优密码子对编码cry6Aa2基因的核酸序列进行密码子优化。去除原序列中存在的poly A和富含AT的ATTTA序列等不稳定元件,并选用大豆偏爱的密码子,使G+C含量从27.24%提高到44.26%。
1.2.2 载体构建
根据cry6Aa2m基因序列设计特异性引物(cry6Aa2m-S:5'GGATCCATGATCATTGACTC 3'cry6Aa2m-AS:5'GAGCTCACTCACTATAGGGC 3'),引物两端分别引入Bam HⅠ和SacⅠ酶切位点。从pEASY-Blunt Simple载体上用高保真酶PCR扩增得到全长基因,扩增片段长度为1 500 bp,反应条件为(25 μL体系):98℃预变性10 min;98℃30 s,55℃15 s,72℃90 s,30个循环;72℃延伸10 min。经双酶切鉴定正确的转化子送交测序,序列完全正确的命名为pEASY-cry6BS。
将pEASY-cry6BS质粒和pCBE-GsGST13表达载体经大量提取与纯化后,分别采用Bam HⅠ、SacⅠ双酶切,分别回收cry6Aa2m小片段和pCBEGsGST13大片段,通过T4DNA连接酶连接,连接产物转化大肠杆菌DH5α感受态细胞,在含有Km的LB培养基平板上筛选转化菌落,Bam HⅠ、SacⅠ双酶切鉴定转化子,经鉴定的重组子命名为pCBE-cry6。
1.2.3 重组载体转化农杆菌
采用冻融法将已构建的植物表达载体pCBE-cry6转化根癌农杆菌EHA105,涂布在含有相应抗生素的YEB培养基上培养,用cry6Aa2m基因特异性引物对转化子进行PCR鉴定。引物和反应体系同1.2.2。
1.2.4 农杆菌介导的子叶节转化法
挑选饱满、无菌斑的大豆种子经氯气灭菌接种在萌发培养基上。萌发5 d后,纵向平均分开2片子叶,去除顶芽和侧芽后在垂直于子叶与下胚轴连接处的有效分化部位轻划5~7刀;将制备好的子叶节外植体置于菌液中,侵染30 min后在22℃进行3 d共培养;转入不定芽诱导培养基;14 d后转入含2 mg·L-1Glufasinate的芽诱导培养基中进行筛选培养;经过28 d的不定芽诱导后,将外植体接入伸长培养基继续培养;待不定芽伸长到3~5 cm后转至生根培养基中,根长到2~3 cm时,移栽到混合土(蛭石/珍珠岩/黑钙土(或黑土)=1/1/1),散射光下覆膜保湿培养,约3~5 d后转入正常光下培养[13]。
1.2.5 PCR检测
取抗性植株叶片,用DNA提取试剂盒(Easy pure Plant Genomic DNA Extraction Kit)提取基因组DNA;用bar基因特异性引物(S:5'TGCACCATCG TCAACCACTACATCG 3',AS:5'CCAGCTGCCAG AAACCCACGTCATG 3')进行PCR检测,扩增片段长度为450 bp,反应参数为:94℃预变性10 min;94℃30 s,63℃30 s,72℃35 s,35个循环;72℃延伸10 min。25 μL PCR反应体系,含有2.5 μL 10倍的PCR Buffer,2 μL 2 mmol·L-1的dNTP Mixture,1 μL 10 μmol·L-1的引物,1 μL的模板,0.25 μL 5 U·μL-1的rTaq聚合酶。
1.2.6 PT-PCR检测
取大豆T1代PCR阳性植株叶片,用RNA prep Plant Kit试剂盒提取样品总RNA,纯化后,以Super ScriptTMⅢReverse Transcriptase试剂盒进行反转录,应用cry6Aa2m基因特异性引物(S:5' AGACTGGATTGGGTTTGCCTG 3',AS:5'CACCA GAGCAACCTTGAAACCG 3')进行PCR检测,扩增片段长度为210 bp,反应条件为:94℃预变性10 min;94℃30 s,50℃30 s,72℃10s,35个循环;72℃延伸10 min。25 μL PCR反应体系,含有2.5 μL 10倍的PCR Buffer,2 μL 2 mmol·L-1的dNTP Mixture,1 μL 10 μmol·L-1的引物,1 μL的模板,0.25 μl 5 U·μL-1的rTaq聚合酶。内参是以18srRNA序列设计的引物(S:5'GGGGGGAGTAT GGTCGCAAG 3',AS:5'TCGCTCCACCAACTAAG AACGG 3'),扩增片段长度约200 bp,反应条件为:94℃预变性10 min;94℃30 s,58℃30 s,72℃20 s,35个循环;72℃延伸10 min。25 μL PCR反应体系,含有2.5 μL 10倍的PCR Buffer,2 μL 2 mmol·L-1的dNTP Mixture,1 μL 10 μmol·L-1的引物,1 μL模板,0.25 μL 5 U·μL-1rTaq聚合酶。
2 结果与分析
2.1 载体构建
从pEASY-Blunt Simple载体上PCR扩增得到cry6Aa2m全长基因,大小1 500 bp,如图2所示。

图2 cry6Aa2m基因的PCR克隆Fig.2 PCR amplification of full lengthcry6Aa2m gene
克隆到的cry6Aa2m全长基因PCR产物连接到pEASY-Blunt cloning kit载体后经Bam HⅠ和SacⅠ双酶切鉴定,切出4 000和1 500 bp两条带,测全长序列,经比对后序列完全正确,说明完全正确的cry6Aa2m全长基因已连接到克隆载体pEASY-cry6BS上。用Bam HⅠ和SacⅠ双酶切pEASY-cry6BS质粒和pCBE-GsGST13表达载体后,将pEASY-cry6BS质粒上的cry6Aa2m小片段和pCBEGsGST13表达载体的大片段连接如图3所示,经Bam HⅠ和SacⅠ双酶切鉴定重组子,切出11 300和1 500 bp两条带。证明该质粒确为重组质粒pCBE-cry6,如图4所示。

图3 植物表达载体pCBE-cry6的构建Fig.3 Construction of plant expression vector pCBE-cry6
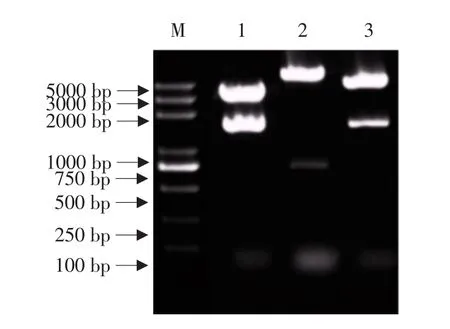
图4 植物表达载体pCBE-cry6的酶切鉴定Fig.4 Characterization with endonucleases of plant expression vector pCBE-cry6
2.2 重组载体转化农杆菌
挑取单菌落为模板,用cry6Aa2m基因特异性引物进行PCR检测。由图5可以看出,泳道3~5均扩增出约1 500 bp条带,证明重组质粒pCBE-cry6已正确导入根癌农杆菌中。
2.3 抗性植株的获得
农杆菌侵染后,将外植体进行共培养(见图6A);3 d后取出洗净转至不定芽诱导培养基(见图6B);14 d后接入含Glufasinate的芽诱导培养基中进行筛选培养(见图6C);28 d的不定芽诱导后,将外植体接入伸长培养基培养(见图6D);待不定芽伸长到3~5 cm时,转至生根培养基中(见图6E);根长至约2~3 cm后,驯化、移栽(见图6F)。最终获得抗性植株44株。
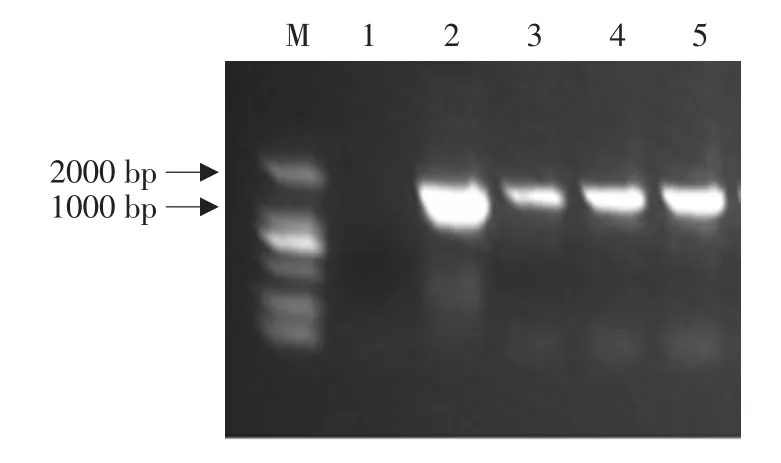
图5 植物表达载体pCBE-cry6转化农杆菌菌落PCR鉴定结果Fig.5 PCR analysis of pCBE-cry6 expression vector transferred into Agrobacterium

图6 大豆遗传转化及再生Fig.6 Genetic transformation and regeneration of soybean
2.4 抗性植株检测
2.4.1 T0代植株PCR检测
分批对约500个子叶节进行侵染后得到44株转pCBE-cry6 T0代大豆品种东农50的抗性植株,以抗性植株的总DNA为模板,pEASY-cry6BS质粒为阳性对照,以水和非转基因基因东农50作为阴性对照,Bar特异序列为引物,进行PCR检测经过三次生物学重复后。其中34株能扩增到450 bp,而阴性对照均无扩增带,初步证明cry6Aa2m基因已整合到大豆基因组中,转pCBE-cry6 T0代大豆品种东农50抗性植株的PCR阳性率达到77.3%(PCR阳性率=PCR阳性植株数/抗性植株数)。PCR结果见图7。
2.4.2 T1代植株的RT-PCR检测
以T1代34株PCR阳性植株总RNA分别为模板,经反转录后得到cDNA后,进行RT-PCR检测,18株PCR阳性植株均能扩增约210 bp的特异条带,证明cry6Aa2m基因在大豆中正常转录。RT-PCR阳性率达到了40.9%(RT-PCR阳性率=RT-PCR阳性植株数/抗性植株数)。RT-PCR电泳结果见图8。
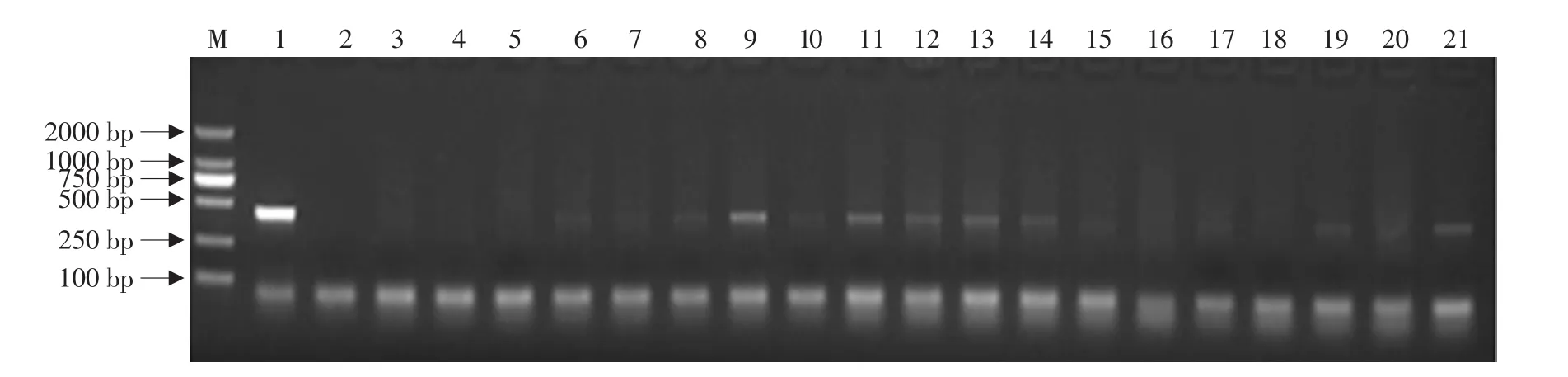
图7 抗性植株PCR检测Fig.7 PCR detection of Glufasinate resistant plants

图8 T1代植株RT-PCR检测Fig.8 RT-PCR detection of T1plants
3 讨论与结论
本研究选用导入抗性基因方式进行抗线虫育种。cry6Aa2基因其编码的晶体蛋白对线虫有很高的杀虫活性,与其高度相似的cry6Aa1基因已在番茄中验证具有杀线虫能力,所以将功能确定的cry6Aa2基因转入大豆,对转基因后代进行分子生物学检测和抗虫检测,获得抗虫、可稳定遗传的转基因纯合体植株,对大豆胞囊线虫的防治、大豆产量和品质提高有重大意义。cry6Aa2基因来自苏云金芽胞杆菌,直接转入高等植物中效果不理想。因为最初利用野生型Bt基因获得的转基因植株抗虫能力都很弱,表达的毒蛋白只占总蛋白的0.1001%或者几乎检测不到。进一步研究发现Bt基因由于是原核基因,在植物中mRNA不稳定,进行翻译时由于某些种类tRNA含量过少而降低了翻译效率。Zou等将Bt毒蛋白基因转入棉花,先将富含AT(63%)密码子的cryIA(b)和cryIA(c)型毒蛋白基因的21%的密码子改造成植物偏爱的同义密码子,使G+C含量从37%提高到49%,最终使外源基因在棉花中的表达量分别提高10倍和100倍[14]。本研究将cry6Aa2m基因在不改变氨基酸序列的前提下选用大豆偏爱的密码子,通过人工改造重新合成基因并结合使用强启动子,有望提高毒蛋白的表达水平,从而提高转基因大豆抗线虫能力。
本研究对已转质粒pCBE-cry6的抗性植株进行PCR检测RT-PCR检测,获得cry6Aa2m基因已经整合到大豆基因组中并进行转录的转基因植株,为进一步确定cry6Aa2m基因插入植物基因组的拷贝数,需进行Southern Blot检测,进一步确定cry6Aa2m基因是否可以在植物中正常表达,进行Western Blot检测。本研究根据大豆密码子的偏爱性,人工合成杀胞囊线虫最小毒力区间cry6Aa2m基因,以pCAMBIA3300为骨架,构建由组成型强启动子E12 CaMV35S调控cry6Aa2m基因、筛选标记基因为除草剂抗性Bar基因的植物表达载体pCBE-cry6,采用已经建立的高效稳定农杆菌介导的大豆子叶节再生体系,对东农50进行遗传转化。本研究侵染子叶节约500个,最终获得抗性苗44株,PCR阳性植株34株,PCR阳性率达到77.3%(PCR阳性率=PCR阳性植株数/抗性植株数),RT-PCR阳性植株18株,RT-PCR阳性率达到40.9%(RT-PCR阳性率=RT-PCR阳性植株数/抗性植株数)。
[1]Konanani B L,Kassim A K,et al.Soil microbial and nematode communities as affected by glyphosate and tillage practices in a glyphosate-resistant cropping system[J].Weed Science,2005,53 (4):536-545.
[2]Qaim M,Zilberman D.Yield effects of genetically modified crops in developing countries[J].Science,2003,299(5608):900-902.
[3]李海燕,朱延明,马凤鸣.植物抗虫基因工程的研究进展[J].东北农业大学学报,2000,31(4):399-405.
[4]吴洪福,郭淑元,李海涛,等.苏云金芽孢杆菌杀虫晶体蛋白结构和功能研究进展[J].东北农业大学学报,2009,40(2):118-122.
[5]余子全,周数,孙明,等.苏云金芽胞杆菌防治植物寄生线虫的研究进展[J].植物保护学报,2004,31(4):418-424.
[6]Li X Q,Wei J Z,Tan A,et al.Resistance to root-knot nematode in tolnato roots expressing a nematieidal Bacillus thuringiensiscrystal protein[J].Plant Biotechno I,2007,5(4):455-464.
[7]Li X Q,Tan A,Voegtline M.Expression ofcry5B protein from Bacillus thuringiensis in plant roots confers resistance to rootknot nematode[J].Biological Control,2008,47(1):97-102.
[8]余子全.苏云金芽胞杆菌杀线虫蛋白基因cry6Aa的表达负调控及杀线虫晶体蛋白进入根结线虫体内的模式[D].武汉:华中农业大学,2008.
[9]郭素霞.苏云金芽胞杆菌菌株YBT-1518新型杀线虫晶体蛋白基因分离的新策略[D].武汉:华中农业大学,2008.
[10]Zhang Z Y,Xiang A Q,Staswick Q.The use of glufosinate ammonium as a selective agent in Agrobacterium-mediated transformation of soybean[J].Plant Cell,Tissue and Organ Culture,1999,56 (8):37-46.
[11]Paz M M,Martinez J C,Kalvig A B,et al.Improved cotyledonarynode method using an alternative explants derived from matureseed for efficient Agrobacterium-mediated soybean transformation [J].Plant Cell Reports,2006,25(3):206-213.
[12]Hfgen R,Willmitzer L.Storage of competent cells for Agrobacterium tumefaciens[J].Nucleic Acids Research,1988,16(20):98-77.
[13]赵晓雯,吴芳芳,狄少康,等.农杆菌介导的大豆子叶节遗传转化技术流程及操作要点[J].大豆科学,2011,30(3):362-368.
[14]Zou Y M,Shi J S,Zhu G Q,et al.Reacting the silencing genes in the transgenic plants[J].Mol Plant Breed,2006,4(1):95-102.
Construction of plant expression vector ofcry6Aa2mgene and transformation intoGlycine max
JI Wei,YANG Likun,BAI Xi,ZHU Yanming,CAI Hua
(School of Life Sciences,Northeast Agricultural University,Harbin 150030,China)
Bacillus thuringiensis(Bt)crystal(cry)proteins are present in thecry6Aa2 intoxicate free-living nematodes.As bacterial codon usage is incompatible with expression in plants,the paper codon-modifiedcry6Aa2 genes designed by selecting the optimal codon usage for each amino acid based on the soybean genome information.The expression vector pCBE-cry6 was constructed.In the vector,cry6Aa2mgene was driven by promoter E12 CaMV35S.Bar served as selection marker andcry6Aa2mgene was transformed into Dongnong50 viaAgrobacterium-mediated method.Total 18 soybean transgenic resistant lines were acquired.PCR and RT-PCR detection indicated that thecry6Aa2mwas successfully integrated into soybean genome and expressed.The research materials yielded in this study might be useful on study of resistence toHeterodera glycines.
soybean;cry6Aa2mgene;genetic transformation;Agrobacterium-mediated method; transformed plant
S572
A
1005-9369(2014)01-0059-06
2012-02-14
国家转基因生物新品种培育重大专项(2009ZX08009-032B)
纪巍(1982-),女,副教授,博士,研究方向为植物基因工程与分子生物学。E-mail:iwei-ji@neau.edu.cn
时间2014-1-9 19:30:59[URL]http://www.cnki.net/kcms/detail/23.1391.S.20140109.1930.001.html
纪巍,杨丽坤,柏锡,等.cry6Aa2m基因植物表达载体构建及大豆的遗传转化[J].东北农业大学学报,2014,45(1):59-64.
Ji Wei,Yang Likun,Bai Xi,et al.Construction of plant expression vector ofcry6Aa2mgene and transformation intoGlycine max[J].Journal of Northeast Agricultural University,2014,45(1):59-64.(in Chinese with English abstract)
