基于高通量测序的海滨雀稗转录组学研究
2014-01-02贾新平叶晓青梁丽建邓衍明孙晓波佘建明
贾新平,叶晓青,梁丽建,邓衍明,孙晓波,佘建明
(江苏省农业科学院农业生物技术研究所 江苏省农业生物学重点实验室,江苏 南京210014)
海滨雀稗(Paspalumvaginatum,Seashore paspalum)是禾本科(Gramineae)黍族(Paniceae)雀稗属(Paspalum)的多年生草本植物,原产于美洲,为潮间带草滩植被的主要组分[1-2]。海滨雀稗是目前耐盐能力最强的草坪草种,还具有较强的耐涝、耐旱、耐践踏和耐磨损特性,能在复杂的逆境条件下生长[3-8]。海滨雀稗的叶色翠绿,景观效果优于狗牙根(Cynodondactylon)、结缕草(Zoysiajaponica)和假俭草(Eremochloaophiuroides)等暖季型禾草,作为草坪草在世界热带与亚热带地区广为种植,已成为21世纪最具发展潜力的草坪草种[9]。同时,海滨雀稗还具有良好的适口性和营养价值,可作为优良牧草加以利用[10]。关于海滨雀稗的遗传多样性、分子标记辅助育种等研究已有报道[11-12],但其基因组和转录组信息的缺乏,造成海滨雀稗分子标记开发、遗传图谱构建、生长发育及其抗逆机理方面的研究相对滞后[13]。
近年来,包括基因组、转录组、蛋白质组等各种组学技术在揭示细胞生理活动规律和生物代谢机理的研究中起着越来越重要的作用,而转录组学是率先发展起来以及应用最为广泛的技术[14]。转录组(transcriptome)是指细胞在特定状态下全部表达的RNA的总和,反映相同基因在不同条件下表达水平的差异,并能揭示不同基因的相互作用及各自功能[15]。转录组测序能全面快速地获得某一物种特定细胞或组织在某一状态下的基因表达情况,用于研究基因结构和功能、可变剪接和新转录本预测等[16-20]。相对于传统的芯片杂交平台,转录组测序无需已知序列设计探针,可对任意物种的整体转录活动进行检测,提供更精确的数字化信号,更高的检测通量以及更广泛的检测范围。对于许多缺乏基因组信息的物种而言,转录组研究已在非模式植物中得到了广泛应用[21-26]。
尽管海滨雀稗具有很高的经济价值和生态价值,但其分子生物学研究进展缓慢,基因数据库资源也十分匮乏。随着高通量测序技术的迅速发展,极大地促进了植物基因表达研究,这样不仅可降低测序的成本和时间,而且还可以获得丰富的数据,有利于植物生长发育及其抗逆等方面的研究[27]。到目前为止,利用新一代高通量测序技术进行草坪种质资源创新与开发的研究还未见报道。本研究首次将Illumina HiSeq 2000高通量测序技术应用到草坪草转录组研究中,将测序得到的海量数据进行拼接与组装,结合生物信息学方法对所获得的unigene进行基因功能注释、功能分类、代谢途径等分析,从功能基因组水平上研究海滨雀稗生长发育过程中重要基因的表达,同时也为进一步的分子标记开发和基因功能研究奠定基础。
1 材料与方法
1.1 试验材料
试验材料为海滨雀稗品种“Adalayd”,由江苏省农业科学院生物技术所植物细胞工程课题组提供。试验于2013年8月在江苏省农业科学院生物技术所温室中进行,选取长势良好、健康的植株叶片,迅速将其放入纸带内,立即经液氮速冻后保存于实验室超低温冰箱中备用。
1.2 RNA提取
利用TRIzoL法提取试验材料海滨雀稗叶片的总RNA。将样品放入研钵,加适量液氮迅速研磨,转移到经DEPC处理的2mL离心管中,加入1mL TRIzoL(Invitrogen,CarLsbad-CA92008,USA),旋涡振荡混匀,室温静置10min;4℃、12000r/min离心10min,将上清液移入新的1.5mL离心管,加入等体积的氯仿∶异戊醇(24∶1)混合液,用力摇15s,室温静置10min;4℃、12000r/min离心10min,将上清液移至新的1.5mL离心管,加入等体积的异丙醇,室温静置10min;4℃、12000r/min离心15min,去除上清液,用RNase-free水配制的75%乙醇洗涤;4℃、12000r/min离心15min,去除上清液;置于冰上自然干燥5min,用RNase-free水溶解,保存于-70℃备用。用AgiLent 2100BioanaLyzer检测RNA提取质量,RIN值≥7.0。
1.3 转录组测序及数据组装
提取样品总RNA后,用带有Oligo(dT)的磁珠富集mRNA。首先加入破碎缓冲液将mRNA打断成短序列,以这些短序列为模板,用六碱基随机引物合成第1条cDNA链,然后加入缓冲液、dNTPs、RNase H和DNA polymerase I合成第2条cDNA链。cDNA经过试剂盒纯化并加EB缓冲液洗脱之后做末端修复、加poly(A)并连接测序接头,然后用琼脂糖凝胶电泳进行序列大小选择,最后进行PCR扩增,建好的测序文库用Illumina HiSeqTM2000进行测序。采用Illumina两个末端(PE)测序法,最初的原始序列为100bp。原始的测序结果去除制备文库时产生的接头序列、两端低质量序列和低度复杂序列,再利用SOAPdenovo软件进行序列拼接[26],之后通过连接两末端和填补空位,将拼接成的重叠群(Contig),进一步组装成unigene。转录组测序产生的原始序列信息已提交到NCBI的SRA数据库(SRA BioProject:SRX383837)。
1.4 功能注释、分类和代谢途径分析
采用序列比对的方法对unigene进行序列相似性分析,使用BLAST程序将拼接得到的unigene与核酸、蛋白质数据库进行比对(E值≤1×10-10),选取最佳的功能注释。核酸数据库为NCBI的非冗余核酸序列数据库(non-redundant nucleotide database,Nt),蛋白质数据库包括 NCBI的非冗余核酸数据库(non-redundant protein database,Nr)和SwissProt(swissprot protein sequence database)蛋白质序列数据库。根据 NCBI数据库的功能注释信息,使用Blast2GO软件[27]得到unigene的GO条目,然后用WEGO软件[28]对所有的unigene进行GO功能分类统计。然后对unigene分别进行蛋白质直系同源数据库(cluster of orthologous groups,COG)功能分类和京东基因与基金组百科全书(kyoto encyclopedia of genes and genomes,KEGG)代谢途径分析。
1.5 SSR位点搜索及分析
对海滨雀稗转录组中的unigene序列进行简单重复序列(simple sequence repeats,SSR)位点搜索,搜索标准为:单、二、三、四、五、六核苷酸基序(motif)至少重复次数分别为10,8,5,4,3,3,对查找的SSR类型进行特征分析。
2 结果与分析
2.1 海滨雀稗转录组数据的组装
采用Illumina HiSeq 2000高通量测序技术对海滨雀稗叶片转录组进行了测序,共得到47520544个reads片段,每个reads的长度为100bp,即测序获得了4752054400bp(4.75Gb)的序列信息。采用SOAPdenovo软件对reads序列聚类进行拼接,共获得966165个contig序列。其中,长度50~100bp的contig序列有761498个,占总体的78.82%;100~200bp的contig序列有125173个,占总体的12.96%;而≥200bp的contig序列有79494个,占总体的8.22%(表1)。由此可见,contig序列主要以长度为50~100bp为主,完全符合Illumina测序的预期结果,为后续的数据组装提供很好的原始数据。
在contig数据的基础上,进一步对序列进行组装,共获得81220个unigene,序列信息达到了87542503bp(87.54Mb),序列大小为201~16328bp,平均长度为1077bp,N50为1680bp。其中,长度200~500bp的unigene有29325个,占总体的36.11%;500~1000bp的unigene有18341个,占总体的22.58%;≥1000bp的unigene有33554个,占总体的41.31%(表2)。

表1 海滨雀稗转录组contig数据组装质量统计Table 1 Data assembly for contig in the transcriptome of P. vaginatum

表2 海滨雀稗转录组unigene数据组装质量统计Table 2 Data assembly for unigene in the transcriptome of P. vaginatum

表3 海滨雀稗unigene的GC含量统计Table 3 Data assembly for GC content of P. vaginatumunigene
GC含量是基因组碱基序列的重要特征之一,能反映基因的结构、功能和进化信息,GC分布不均匀导致基因组不同GC含量序列其性质和功能也有差异。海滨雀稗unigene的GC平均含量为49.98%,其中GC含量40%~60%的unigene(59903个)占总体的73.75%,GC含量20%~40%的unigene(6552个)占总体的8.07%,GC含量60%~80%的unigene(14765个)占总体的18.18%,而GC含量过高(大于80%)或过低(小于20%)的unigene不存在,表明GC含量基本呈正态分布(表3)。
用RPKM方法计算unigene的表达水平,可消除基因长度差异和测序深度的影响[29]。海滨雀稗全部unigene的RPKM平均值为28.68,最大值为50812.5(unigene 42358)。281个unigene的RPKM 值大于500,其中许多基因参与到海滨雀稗的多种生理活动和代谢过程中。91个unigene的RPKM值低于0.2,说明Illumina HiSeq 2000能够检测到极低水平的基因表达。
2.2 unigene的功能注释、分类和代谢途径分析
2.2.1 unigene的序列相似性分析 使用BLAST程序将组装得到的unigene与Nr、Nt、SwissProt数据库进行比对,进行unigene的序列相似性分析。结果表明,38446个unigene在Nr数据库中可找到相似序列,E值小于1×10-100的unigene有22629个(占总体的58.86%),E值介于1×10-10~1×10-100的unigene有15817个(占总体的41.14%);相似序列匹配的近缘物种中,高粱(Sorghumbicolor)所占比例最高(45.76%),随后依次是玉米(Zeamays,38.68%)、水稻(Oryzasativa,8.91%)、小麦(Triticumaestivum,2.45%)和其他物种(4.20%)(图1)。46169个unigene在Nt数据库中可找到相似序列,E值小于1×10-100的unigene有28599个(占总体的61.94%),E值介于1×10-10~1×10-100的unigene有17570个(占总体的38.06%);相似序列匹配的近缘物种中,高粱所占比例最高(62.72%),随后依次是玉米(25.82%)、水稻(3.81%)、短柄草(Brachypodiumdistachyon,2.11%)和其他物种(5.54%)。24471个unigene在SwissProt数据库中可找到相似序列,E值小于1×10-100的unigene有8742个(占总体的35.72%),E值介于1×10-10~1×10-100的unigene有15729个(占总体的64.28%);相似序列匹配的近缘物种中,拟南芥(Arabidopsisthaliana)所占比例最高(46.03%),随后依次是水稻(20.10%)、玉米(8.30%)、小麦(3.73%)和其他物种(21.84%)(图1)。由于缺乏海滨雀稗的基因组、EST和蛋白序列信息,部分unigene在数据库中无法匹配到已知基因。

图1 海滨雀稗unigene的序列相似性分析Fig.1 Characteristics of homology search of P. vaginatumunigene
2.2.2 unigene的GO分类 基因本体论(gene ontology,GO)是一个国际标准化的基因功能分类数据库,用于全面地描述不同生物中基因的生物学特征。结合GO数据库对海滨雀稗的unigene进行功能分类,从宏观上认识海滨雀稗表达基因的功能分布特征。GO数据库包括3个相对独立的本体,分别描述所处的细胞组分(cellular component)、分子功能(molecular function)和参与的生物学过程(biological process)。研究结果表明,可将海滨雀稗unigene划分为48个功能组,并对每个功能组涉及的unigene进行了统计分析。从图2中可以看出,51497个unigene归属于细胞组分,22718个unigene归属于分子功能,60856个unigene归属于生物学过程,这一分类结果显示了海滨雀稗生长过程中基因表达谱的总体情况。其中,“细胞成分”(18763个)、“细胞进程”(17347个)、“代谢进程”(13891个)和“结合活性”(10726个)功能组中涉及的unigene较多,而“翻译调节活性”(7个)、“金属伴侣蛋白活性”(4个)、“氮素利用”(2个)和“蛋白标签”(2个)功能组中涉及的unigene较少。
2.2.3 unigene的COG功能分类 蛋白质直系同源数据库(cluster of orthologous groups,COG)是对基因产物进行直系同源分类的数据库。将海滨雀稗unigene与COG数据库进行比对,预测unigene功能并进行分类统计。研究结果表明,海滨雀稗unigene根据其功能大致可分为25类,并对每类的unigene进行了统计分析(图3中用A~Z表示)。从图中可以看出,unigene涉及的COG功能类别比较全面,涉及了大多数的生命活动。其中,一般功能预测类基因最多(4716个);其次是未知功能类基因(3306个)、翻译,核糖体结构和生物发生类基因(2662个)、翻译后修饰,蛋白质折叠和分子伴侣类基因(2247个)和碳水化合物运输和代谢类基因(2087个);而胞外结构类基因(11个)和核结构类基因较少(7个);其他类别的基因表达丰度都各不相同。

图2 海滨雀稗unigene的GO分类Fig.2 GO functional categories of P. vaginatumunigene
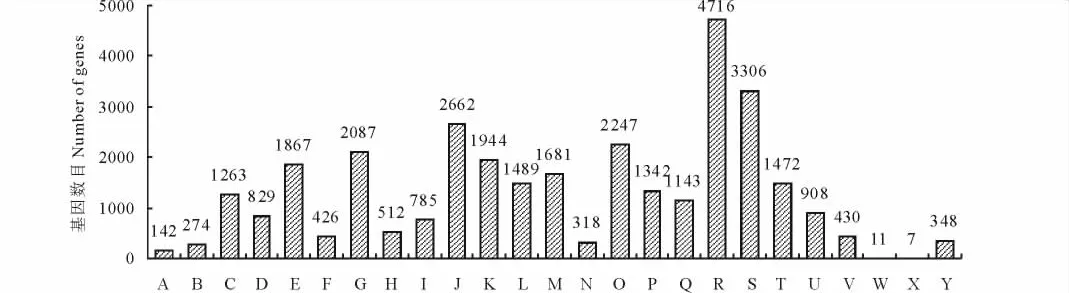
图3 海滨雀稗unigene的COG功能分类Fig.3 COG function classification of P. vaginatumunigene
2.2.4 unigene的 KEGG分析 KEGG(kyoto encyclopedia of genes and genomes)是系统分析基因产物在细胞中的代谢途径以及基因产物功能的数据库。根据KEGG数据库的注释信息能进一步得到unigene的pathway注释[30]。结合KEGG数据库,对海滨雀稗的unigene可能参与或涉及的代谢途径进行了统计分析。研究结果表明,可将海滨雀稗的unigene归属于五大类的代谢途径,主要包括碳水化合物代谢、氨基酸代谢、脂类物质代谢、次生物质代谢、复制与修复、转录与翻译、信号转导等19类代谢途径(图4)。将KEGG pathway数据库作为参考,可将unigene定位到112个具体的代谢途径分支。其中,涉及糖异生和糖酵解途径的基因有140个,占总体的3.35%;磷脂酰肌醇信号系统途径的基因有133个,占总体的3.18%;甘油磷脂代谢途径的基因有130个,占总体的3.11%;苯丙氨酸代谢途径的基因有79个,占总体的1.89%;RNA降解途径的基因有68个,占总体的1.63%;黄酮类化合物合成途径的基因有44个,占总体的1.05%;植物与病原物互作的基因有23个,占总体的0.56%(表4)。
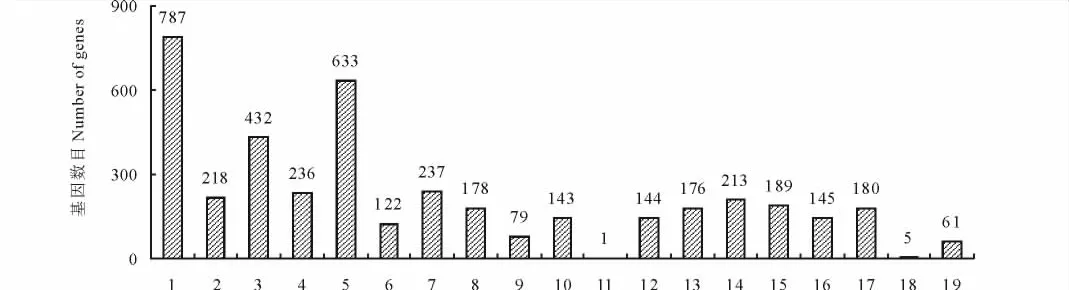
图4 海滨雀稗unigene的KEGG分类Fig.4 KEGG classification of P. vaginatumunigene
2.3 SSR分析
对海滨雀稗的81220个unigene进行SSR位点搜索,共检测到22721个SSR位点。SSR的类型丰富,单核苷酸至六核苷酸重复类型均存在,所占比例变化较大(表5)。其中,三核苷酸重复所占比例最高,达到了31.71%;比例最低的是二核苷酸重复,仅为4.35%;五核苷酸重复和六核苷酸重复所占比例基本相同,分别为14.15%和14.67%。在检测到的SSR中,出现频率最高的10类基序为:A/T(6198个)、CCG/CGG(2992个)、AGC/CTG(1277个)、AGG/CCT(1249个)、AG/CT(776个)、ACG/CGT(516个)、AAG/CTT(415个)、ACC/GGT(376个)、AGAGG/CCTCT(259个)、AGGG/CCCT(197个)。上述SSR特征分析,有助于开展海滨雀稗及其同属物种的基因组差异分析、通用性标记开发和遗传图谱构建的研究。
3 讨论
随着新一代高通量测序技术的广泛应用,植物基因组研究得到快速发展,但草坪草基因组研究还相对较少。Illumina高通量测序的数据量大、速度快、成本低、效率高[31],适合于没有基因组信息的海滨雀稗展开转录组测序研究。基于转录组学在功能基因组学研究中的重要价值,本研究应用Illumina高通量测序技术对海滨雀稗转录组进行测序,研究其基因表达谱和挖掘生长发育过程中的重要表达基因。对海滨雀稗转录组进行测序,获得了
47520544个reads序列;对reads序列进行拼接,共获得了966165个contig序列。在contig数据的基础上,序列组装后得到了81220个unigene,长度大小从201~16328bp,平均长度为1077bp,N50值为1680bp(N50指从组装最长的unigene依次向下求长度的总加和,当累加长度达到组装长度的一半时,对应的unigene长度是N50长度。N50值越大,反映组装得到的长片段越多,组装效果就越好。测序数据产量和数据组装质量是转录组测序完成情况的重要指标。以上研究结果表明,此次序列组装的质量和长度可以满足转录组分析的基本要求,且新一代高通量测序技术是批量发现海滨雀稗功能基因的更为有效手段,进一步说明Illumina HiSeq 2000是高通量转录组测序的可靠平台。

表4 海滨雀稗unigene的代谢途径分析Table 4 Analysis of metabolic pathways of P. vaginatumunigene

续表4 Continued
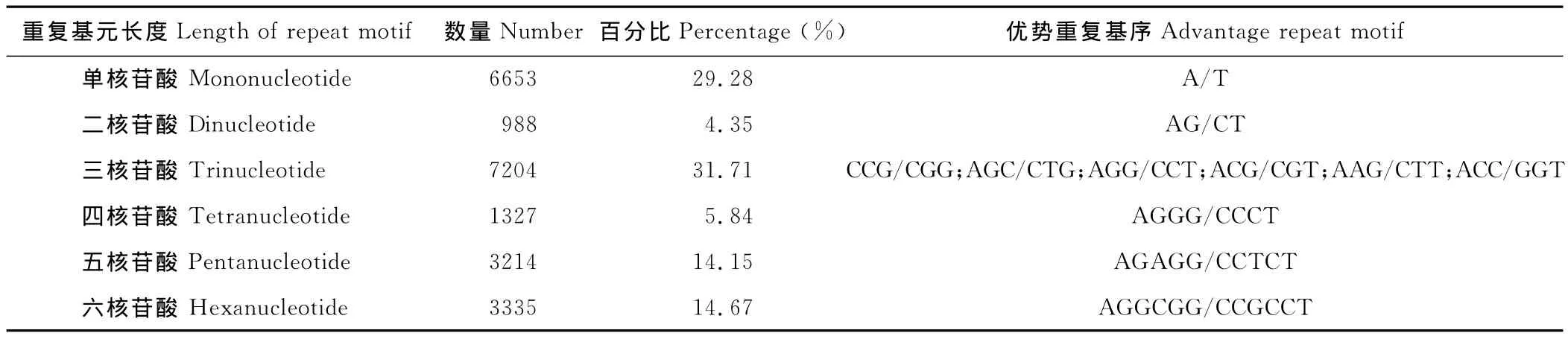
表5 海滨雀稗SSR不同重复基序分布及优势碱基组成Table 5 Distribution and compositions of the dominant repeat of the different repeat motifs for SSR
结合生物信息学分析方法,对海滨雀稗unigene与Nr、Nt、SwissProt数据库进行比对,进行序列相似性和功能注释分析。46169个unigene与其他近缘生物的已知基因具有不同程度的同源性,并且还获得了35051个新的unigene(占总体的56.84%),表明在对海滨雀稗基因组及遗传背景几乎不清楚的情况下,高通量测序技术是批量发现海滨雀稗功能基因的有效手段。虽然GO是个标准化的生物信息本体数据库,被广泛地用于基因的注释功能,然而由于GO结构设计上的缺陷以及基因的许多特征还未被发现,使得这种基因注释信息尚不完全。因此,本研究中的海滨雀稗unigene基于GO数据库进行的相关功能注释信息还不完善,还有部分的unigene没有赋予了可能的GO条目,有待通过其他生物信息学方法对unigene功能注释进一步补充。利用COG数据库对海滨雀稗unigene进行基因功能分类,可从基因组水平上找寻直系同源体,预测未知ORF的生物学功能,可以大大提高基因功能注释的准确性。根据KEGG数据库对上述unigene进行代谢途径分析,涉及112个具体的代谢途径分支,参与到海滨雀稗体内的碳水化合物代谢、脂类代谢、次生物质代谢等过程中,为进一步大量挖掘海滨雀稗生长发育过程中的重要表达基因,开展海滨雀稗的基因克隆及功能验证等研究提供了基础数据。
SSR分子标记具有操作简便、重复性好、多态性丰富、遗传信息量大、共显性遗传等优点,已在遗传多样性分析、遗传图谱构建、功能基因发掘、分子标记辅助育种等研究中得到了广泛应用[32-36]。采取实验室手段开发SSR引物费时,耗力,成本高,试验复杂,基于转录组数据库信息进行SSR分子标记开发将是一种既经济又有效的方法。目前,海滨雀稗可利用的分子标记数量非常有限,转录组产生的海量数据为SSR分子标记的开发提供了更丰富和极有价值的可利用资源。本研究通过查找发现了22721个SSR位点,SSR不但出现频率高,而且类型丰富。利用在线引物设计软件共设计出8758对SSR引物,进一步可对这些SSR引物进行扩增检测,筛选出扩增稳定、条带清晰、多态性好的引物,为进一步开发新的SSR标记奠定了基础。SSR分子标记的开发可用于草坪草功能基因的挖掘、丰富分子标记类型、遗传资源评价、重要性状的辅助选择等研究,有助于促进草坪草遗传育种的发展[37-38]。
本研究首次在国内外采用Illumina HiSeq 2000高通量测序技术建立了海滨雀稗转录组数据库,获得了大量的转录本信息,并对表达基因进行了序列组装、功能注释、代谢途径等分析,为今后更深入研究海滨雀稗功能基因组、基因克隆及抗逆机理研究提供了极大的方便,而且该转录组数据还可以作为今后海滨雀稗基因组的参考序列,为海滨雀稗的分子生物学研究提供宝贵的基因组数据来源。
[1]解新明,卢小良.海雀稗种质资源的优良特性及其利用价值[J].华南农业大学学报,2004,25:64-67.
[2]常盼盼,钟小仙,刘智微.海滨雀稗体细胞突变体SP2008-3的特异性分析[J].草业学报,2012,21(6):207-212.
[3]陈静波,褚晓晴,李珊,等.盐水灌溉对7属11种暖季型草坪草生长的影响及抗盐性差异[J].草业科学,2012,29(8):1185-1192.
[4]卢少云,郭振飞.草坪草逆境生理研究进展[J].草业学报,2003,12(4):7-13.
[5]Cardona C A,Duncan R R,Lindstrom O.Low temperature tolerance assessment in paspalum[J].Crop Science,1997,37(4):1283-1291.
[6]Carrow R N.Seashore paspalum ecotype responses to drought and root limiting stresses[J].USGA Turfgrass and Environmental Research,2005,4(13):1-9.
[7]Huang B,Duncan R R,Carrow R N.Drought resistance mechanisms of seven warm-season turfgrasses under surface soil drying:I.Shoot response[J].Crop Science,1997,37(6):1858-1863.
[8]Unruh,J B,Brecke B J,Partridge D E.Seashore paspalum performance to potable water[J].USGA Turfgrass and Environmental Research,2007,6(23):1-10.
[9]叶晓青,佘建明,梁流芳,等.海雀稗体细胞低温筛选获得耐寒突变体[J].草地学报,2010,18(1):97-102.
[10]刘国道,罗丽娟.中国热带饲用植物资源[M].北京:中国农业大学出版社,1999:176-179.
[11]贺小霞,刘一明,王兆龙.海滨雀稗栽培品种的形态特征与AFLP分子标记分析[J].草地学报,2011,19(1):164-170.
[12]解新明,卢小良,孙雄松,等.海雀稗种质资源RAPD分子标记的遗传多样性研究[J].华南农业大学学报,2004,25:10-15.
[13]井赵斌,魏琳,俞靓,等.转录组测序及其在牧草基因资源发掘中的应用前景[J].草业科学,2012,28(7):1364-1369.
[14]Lockhart D J,Winzeler E A.Genomics,gene express and DNA arrays[J].Nature,2000,405:827-836.
[15]Velculescu V E,Zhang L,Zhou W,etal.Characterization of the yeast transcriptome[J].Cell,1997,88(2):243-251.
[16]Alagna F D,Agostino N,Torchia L,etal.Comparative 454pyrosequencing of transcripts from two olive genotypes during fruit development[J].BMC Genomics,2009,10:399.
[17]Barakat A,DiLoreto D S,Zhang Y,etal.Comparison of the transcriptomes of American chestnut(Castaneadentata)and Chinese chestnut(Castaneamollissima)in response to the chestnut blight infection[J].BMC Plant Biology,2009,9:51.
[18]Dassanayake M,Haas J S,Bohnert H J,etal.Shedding light on an extremophile lifestyle through transcriptomics[J].New Phytologist,2009,183(3):764-775.
[19]Maher C A,Kumar-Sinha C,Cao X,etal.Transcriptome sequencing to detect gene fusions in cancer[J].Nature,2009,458:97-101.
[20]Wang E T,Sandberg R,Luo S,etal.Alternative isoform regulation in human tissue transcriptomes[J].Nature,2008,456:470-476.
[21]张争,高志晖,魏建和,等.三年生白木香机械伤害转录组学研究[J].药学学报,2012,47(8):1106-1110.
[22]杨楠,赵凯歌,陈龙清.蜡梅花转录组数据分析及次生代谢产物合成途径研究[J].北京林业大学学报,2012,34(1):104-107.
[23]Franssen S U,Shrestha R P,Brautigam A,etal.Comprehensive transcriptome analysis of the highly complexPisumsativumgenome using next generation sequencing[J].BMC Genomics,2011,12:227.
[24]Der J P,Barker M S,Wickett N J,etal.De novo characterization of the gametophyte transcriptome in bracken fern,Pteridiumaquilinum[J].BMC Genomics,2011,12(1):99.
[25]王晓锋,何卫龙,蔡卫佳,等.马尾松转录组测序和分析[J].分子植物育种,2013,11(3):385-392.
[26]Li R Q,Zhu H M,Ruan J.De novo assembly of human genomes with massively parallel short read sequencing[J].Genome Research,2010,20(2):265-272.
[27]Conesa A,Gtz S,García-Gómez J M,etal.Blast2GO:a universal tool for annotation,visualization and analysis in functional genomics research[J].Bioinformatics,2005,21(18):3674-3676.
[28]Ye J,Fang L,Zheng H,etal.WEGO:a web tool for plotting GO annotations[J].Nucleic Acids Research,2006,34:293-297.
[29]Mortazavi A,Williams B A,McCue K,etal.Mapping and quantifying mammalian transcriptomes by RNA-Seq[J].Nature Methods,2008,5(7):621-628.
[30]林萍,曹永庆,姚小华,等.普通油茶种子4个发育时期的转录组分析[J].分子植物育种,2011,9(4):498-505.
[31]祁云霞,刘永斌,荣威恒.转录组研究新技术:RNA-Seq及其应用[J].遗传,2011,33(11):1191-1202.
[32]宋建,陈杰,陈火英,等.利用SSR分子标记分析番茄的遗传多样性[J].上海交通大学学报,2006,24(6):524-528.
[33]陈海梅,李林志,卫宪云,等.小麦EST-SSR标记的开发、染色体定位和遗传作图[J].科学通报,2005,50(20):2208-2216.
[34]刘峰,王运生,田雪亮,等.辣椒转录组SSR挖掘及其多态性分析[J].园艺学报,2012,39(1):168-174.
[35]Yang H B,Liu W Y,Kang W H,etal.Development of SNP markers linked to the L locus inCapsicumspp.by a compara-tive genetic analysis[J].Molecular Breeding,2009,24(4):433-446.
[36]Yi G,Lee J M,Lee S,etal.Exploitation of pepper EST-SSRs and an SSR-based linkage map[J].Theoretical and Applied Genetics,2006,114(1):113-130.
[37]曾亮,袁庆华,王方,等.冰草属植物种质资源遗传多样性的ISSR分析[J].草业学报,2013,22(1):260-267.
[38]陈群,袁晓君,何亚丽.高羊茅单株耐热性相关分子标记的筛选及其与越夏性的关系研究[J].草业学报,2013,22(5):84-95.
