后过渡金属催化烯烃聚合稳定性的改进
2013-12-23廖恒,伍青
廖 恒,伍 青
(中山大学 化学与化学工程学院 高分子研究所,广东 广州 510275)
后过渡金属由于容易发生β-H消除,因此在很长一段时间内人们认为它只能用于烯烃齐聚,不能催化烯烃进行聚合反应[1]。同时由于后过渡金属比前过渡金属拥有更多的d轨道电子,因此人们认为它活化烯烃单体的能力较弱[2]。直到20世纪90年代中期,Johnson等[3]研究发现阳离子型后过渡金属的亲电性高,可能会比人们预期的更快速地进行烯烃单体插入,高活性地制备高相对分子质量聚合物。
与前过渡金属催化剂相比,后过渡金属催化剂可以通过调节乙烯压力来合成线型或高度支化甚至超支化的聚乙烯[4],不需α-烯烃与乙烯共聚[5]。而且后过渡金属亲氧性弱,对极性基团容忍性强,容易实现烯烃与极性单体共聚[6]。但后过渡金属催化剂的热稳定性不好,常见的α-二亚胺镍、钯催化剂在较高温度下快速失活[7-8],且随聚合温度的升高,聚合产物的相对分子质量明显下降。后过渡金属催化剂的不稳定性限制了其工业化应用[9]。
本文综述了几种改进后过渡金属催化体系稳定性的方法,简要归纳了影响后过渡金属催化剂稳定性的因素。
1 [N,N]配体催化剂
α-二亚胺镍、钯催化剂(见催化剂1和2)能高活性地催化乙烯聚合,活性(每小时每摩尔Ni生成的聚乙烯的质量)可达107g/(mol·h)[3]。由于后过渡金属中心与增长链β-H发生作用,使催化中心沿增长链迁移,即所谓的“链行走”(Chain walking),最终生成支化聚乙烯。聚合产物的支化度随温度和乙烯压力的不同而不同,温度升高支化度增大,但乙烯压力增大则支化度降低。这种影响对α-二亚胺钯催化剂尤为明显[4]。同时α-二亚胺钯催化剂的中心金属与β-H的相互作用而不易发生链转移反应,可以实现乙烯及α-烯烃的活性聚合[10-11]。
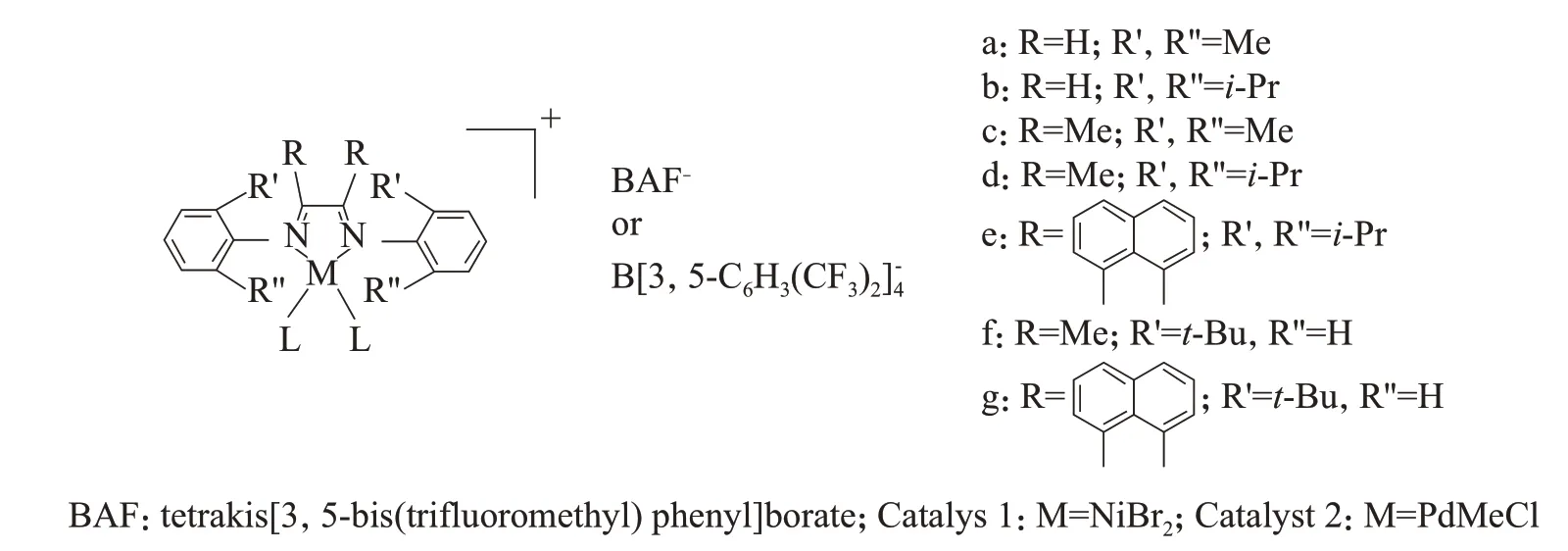
与很多前过渡金属催化剂不同,后过渡金属α-二亚胺催化剂在相对温和的条件下易失活。研究α-二亚胺Pd(Ⅱ)催化剂时发现,当苯胺基通过C—N键自由旋转至与二亚胺金属配位环共平面时,其邻位上的烷基与金属中心相互作用,发生C—H键活化,形成六元环中间体进而失活[8]。增大配体位阻能抑制C—N键的自由旋转,从而抑制C—H键活化。增加给电子体浓度或使用更强的给电子体也能抑制C—H键活化,如以乙腈为给电子体的配合物比以乙醚为给电子体的配合物更稳定。
将苯胺的2,6-位用大体积的苯基取代得到的催化剂3(见图1),在80 ℃及H2存在下,能高活性地催化乙烯聚合[12]。而具有相同配体骨架结构但苯胺2,6-位用异丙基取代的二亚胺催化剂,在H2存在下会快速失活,室温下已无催化活性;在60 ℃时,用三甲基铝活化苯胺2,6-位异丙基取代的二亚胺催化剂,该催化剂在反应开始时显示出常规活性,但迅速失活,3 min内乙烯吸收速率曲线迅速下降到很低水平。但在相同条件下,苯胺2,6-位芳基取代的二亚胺催化剂的吸收速率曲线保持在高水平上[13]。将苯胺2,6-位用大体积的呋喃、苯并呋喃基取代得到的二亚胺镍催化剂4和催化剂5(见图1)也具有很高的热稳定性[14],60℃时,二者均能得到超高相对分子质量的聚乙烯(Mw=3×106g/mol,双峰分布),且在150 ℃时仍有一定的活性,产物的Mw=2.3×104g/mol。
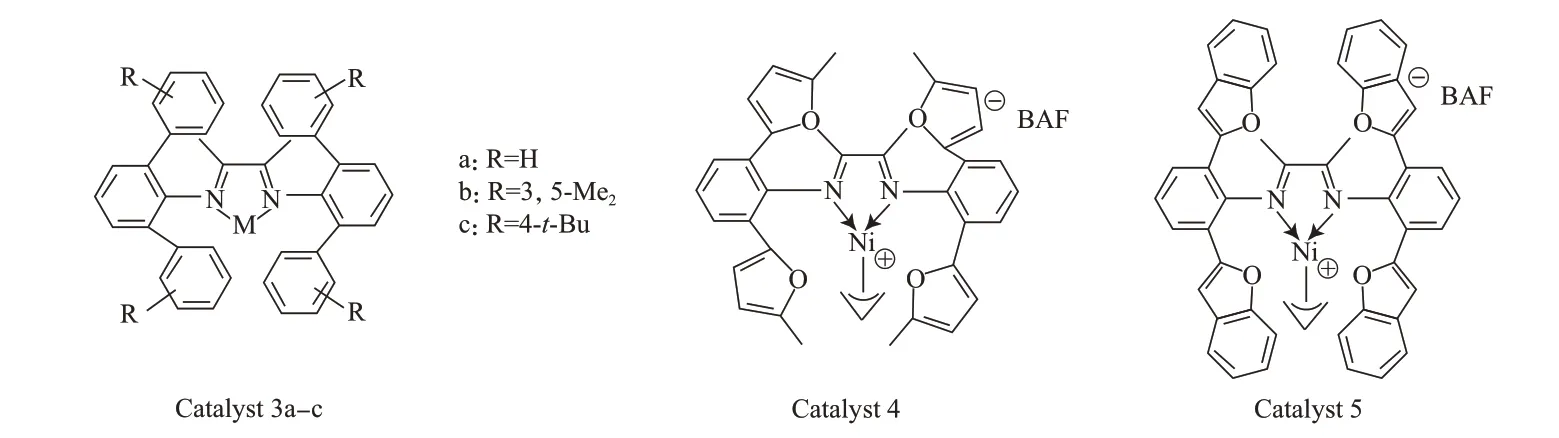
图1 大位阻α-二亚胺催化剂Fig.1 Steric bulky α-diimine catalysts.
环芳型配合物6(见图2)不仅能有效屏蔽轴向单体进攻金属中心而引起链转移反应,而且大环有一定的刚性,能有效防止C—N键的自由旋转,提高催化剂的稳定性[15]。环芳型镍催化剂在90 ℃、改性甲基铝氧烷(MMAO)助催化剂存在下可实现乙烯聚合,且具有很高的活性,转化频率(TOF)达到106h-1级,得到高相对分子质量产物(Mn达到105g/mol级)。
同样,环芳型钯催化剂也具有很好的稳定性。相对于结构相似但非环型的钯催化剂3(常温下即快速分解),环芳型钯催化剂在60 ℃下仍能保持几十小时的活性[16]。

图2 环状配合物及大骨架位阻配合物Fig.2 Cylic and bulky substituted backbone complexes.
与芳环型催化剂相比,烷基桥联的大环催化剂7(见图2)的催化性能欠佳。这可能是由于金属中心轴向上的烷基的空间位阻过大,导致单体与金属中心配位困难,升高了单体插入能垒,使得催化剂活性较低[17]。同时,XRD表征结果显示,烷基上的一些H原子非常靠近金属中心,可能引起C—H键活化,从而使金属中心失活。这说明为提高催化剂的稳定性,在利用大体积基团增大空间位阻的同时,还应避免因基团的增大使其更容易接近金属中心而造成催化剂失活。
二亚胺骨架对催化剂聚合性能也有很大影响[18]。当用甲基取代骨架上的H后,钯催化体系的反应活性和产物相对分子质量均大幅提高;镍催化体系的产物相对分子质量和支化度也有所提高。增大二亚胺骨架上的空间位阻,可抑制C—N键的自由旋转,防止烷基与金属中心作用,降低催化剂分解速率。如对于两个苯胺邻位均为单叔丁基取代的催化剂2,苊基骨架的2g比甲基骨架的2f更易发生C—H键活化,引起催化剂失活[8]。
根据以上骨架空间位阻对催化剂稳定性的影响设计合成的以莰基为骨架的二亚胺镍催化剂8(见图2)显示出良好的热稳定性[19]。在0.05 MPa的乙烯压力下,反应温度从40 ℃升至80 ℃,催化剂活性无明显降低,且在高温下仍可得到高相对分子质量的支化聚乙烯。莰基上的一个甲基靠近金属中心的轴向位置,抑制了轴向单体与金属中心配位引起的链转移反应;另一个甲基位于苯胺基团后面,有效抑制了C—N键的自由旋转,防止C—H键活化引起的金属中心失活,从而保证催化剂具有良好的热稳定性。
电子效应对催化剂性能也有很大影响[20-21]。通过研究催化剂9(见图3)中苯胺对位为具有不同电子效应的取代基时发现,给电子基团能提高催化剂的稳定性并延长其寿命,同时使单体插入的金属中心过渡态更稳定,从而可明显提高产物的相对分子质量。如对位用NMe2(Me为甲基)取代的钯催化剂9g所得产物的相对分子质量比未取代的催化剂9d高一倍[21]。

图3 不同电子效应基团取代的α-二亚胺催化剂Fig.3 α-Diimine catalysts with various electron-donating and withdrawing groups.
研究具有不同电子效应取代基的环芳型镍催化剂10(见图3)发现,该类催化剂能在115 ℃下催化乙烯聚合,它们的活性差别不明显,且都明显低于80 ℃时的活性。但值得注意的是,用具有相同配体的钯催化剂在60 ℃下催化乙烯聚合,当芳胺对位吸电子基为Cl-取代时,该催化剂比其他取代基催化剂具有更好的热稳定性,能持续聚合48 h以上。以上环芳型与非环型催化剂在具有不同电子效应取代基的存在下表现出不同的聚合性能,表明取代基的适当电子效应是保持金属中心稳定的一个重要因素[22]。
与环芳型催化剂相比,苯基邻位F原子取代的催化剂11(见图4)不仅可以得到高相对分子质量、低支化度的聚合物,而且该催化剂具有更好的热稳定性,在105 ℃下70 min内活性降低幅度很小[23]。NMR表征结果显示,苯环上的F原子与金属中心直接相互作用,稳定了金属中心的14e-中间体,抑制了β-H的消除,使得链转移、链行走和催化剂分解都受到抑制,因此催化剂具有很好的热稳定性。
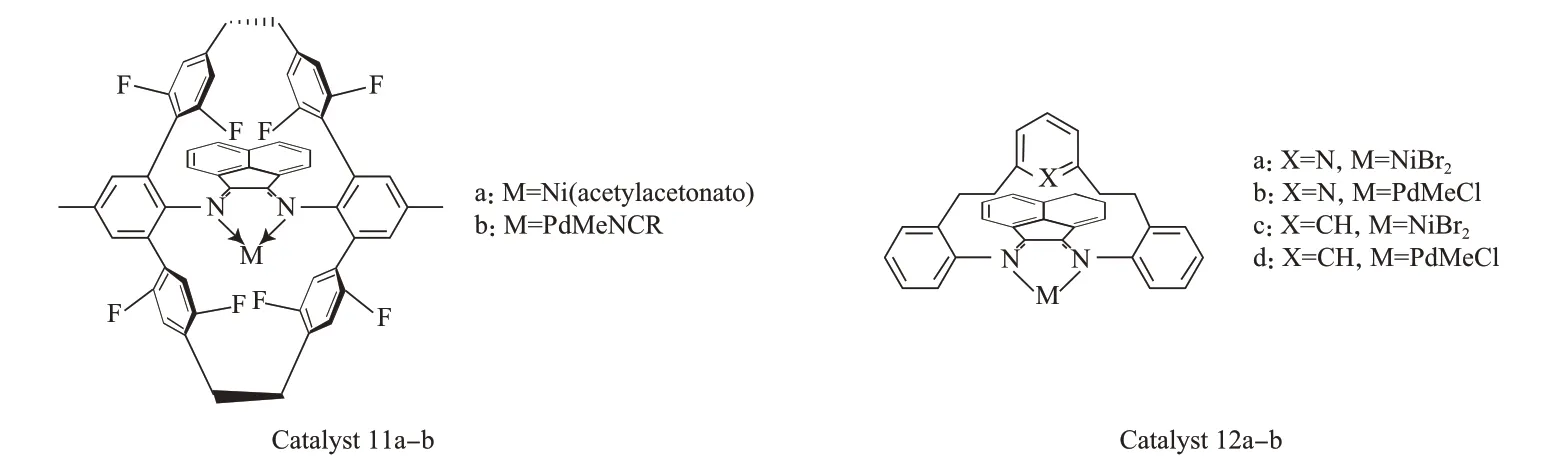
图4 氟代环芳和轴向给电子配合物Fig.4 Fluorinated cyclophane and axial donating complexes.
半环型催化剂12a和12b(见图4)的轴向基团可以可逆地与金属中心作用,从轴向上为14e-金属中心提供电子使其稳定,抑制β-H消除,从而阻止链转移反应的发生,达到暂时保护反应中间体的目的[24]。实验结果表明,轴向基团可使吡啶基取代的催化剂催化聚合得到更高相对分子质量及低支化度的线型结晶聚合物。
2-胺基吡啶的镍配合物13(见图5)可在甲基铝氧烷(MAO)助催化作用下引发乙烯聚合,但其催化活性低且产物中低聚物的含量高[25]。在吡啶环与胺基间的桥接碳上进行适当取代后得到的配合物14(见图5)可以制得高相对分子质量的乙烯聚合物。当R为2,4,6-三甲基苯基时,催化剂能在-10℃下催化乙烯聚合,且在长达6 h内聚合物的相对分子质量随时间的延长呈线性增加,相对分子质量分布在1.2以内[26]。

图5 吡啶胺基和胺基亚胺配合物Fig.5 Aminopyridine and amine-imine complexes.
以MAO为助催化剂,在25 ℃、2.8 MPa乙烯压力下,胺基亚胺催化剂15a(见图5)能以较高活性催化乙烯聚合,得到支化聚乙烯。增大苯胺邻位取代基可降低聚合产物的相对分子质量,使相对分子质量分布变窄[27-28]。催化剂16(见图5)能催化乙烯聚合,得到相对分子质量分布窄的高支化度聚合物[29]。20 ℃时,苯胺2,6-位取代基的位阻越大,产物的相对分子质量越高,相对分子质量分布越窄。催化剂16a可在一氯二乙基铝助催化、50 ℃下实现乙烯活性聚合,聚合物的相对分子质量分布为1.08;35 ℃时,该催化剂能保持长达120 min的活性聚合特征(聚合物相对分子质量分布小于1.10),没有失活现象。
2 吡啶二亚胺催化剂
1998年,文献[30]和[31]分别报道了吡啶二亚胺铁、钴催化剂(见图6催化剂17)。在该类催化剂中,亚胺芳环近似垂直于吡啶环,配位的3个N原子处于纬线位置,而Cl原子处于经线位置。吡啶二亚胺铁配合物能高活性催化乙烯聚合,活性可达108g/(mol·h·MPa),得到高度线型、高相对分子质量的聚合物,其Mw超过105g/mol。相对于吡啶二亚胺铁配合物,吡啶二亚胺钴配合物催化乙烯聚合的活性较低,一般低一个数量级。
由于催化体系中存在向单体和向烷基铝助催化剂转移的两种链转移方式,因此产物中高聚物和低聚物共存,相对分子质量分布宽。与催化剂17b相比,具有不对称大位阻的苯胺取代基吡啶二亚胺铁催化剂18b(见图6)具有更好的热稳定性,产物相对分子质量更高[32]。在30 ℃下,虽然两种催化剂制得的产物的GPC曲线都呈双峰分布,但催化剂18b在高相对分子质量部分峰值处的相对分子质量为催化剂17b的1.5倍,且前者产物中高相对分子质量部分所占的比例也比后者高。经实验发现,催化剂18b在70 ℃时的活性(相对于1 mol的Fe)最高,为4.4×107g/(mol·h),且聚合产物为线型聚乙烯(熔点为136 ℃)。而催化剂17b在50 ℃时活性达到最高,为1.3×107g/(mol·h)(相对于1 mol的Fe)。70 ℃时,与催化剂17b相比,催化剂18b的活性保持得更好,半衰期达到25 min,而催化剂17b的半衰期仅为10 min。同样具有苯胺基团大位阻的催化剂19(见图6)具有良好的催化性能。在MAO助催化作用下,当乙烯压力为1.0 MPa时,催化剂19c在60℃时乙烯聚合活性最高;而在MMAO助催化作用下,催化剂19c在80 ℃时乙烯聚合活性最高,为107g/(mol·h)级,得到高相对分子质量聚合物(Mw达2.95×105g/mol),且产物中没有低聚物[33]。

图6 吡啶二亚胺配合物Fig.6 Bis(imino)pyridyl complexes.
亚甲基桥联双核吡啶二亚胺铁催化剂20b(见图7)在0 ℃时活性能在20 min内保持平稳且随时间的延长活性缓慢降低,而非桥联的吡啶二亚胺铁催化剂17b具有很高的初始活性,但失活快,这表明桥联后的双核催化剂能有效抑制活性中心的失活[34]。

图7 双核及三核吡啶二亚胺配合物Fig.7 Binuclear and trinuclear bis(imino) pyridyl complexes.
将3个吡啶二亚胺单体用烷基桥联,形成的大环骨架可有效抑制链转移反应的发生,而且能保护活性金属中心,提高催化剂的热稳定性[35]。实验结果表明,在0 ℃、0.1 MPa乙烯压力和MMAO助催化作用下,大环型催化剂21(见图7)的聚合产物的GPC曲线呈单峰特性(仅有微弱肩峰),且产物相对分子质量较高(Mw=4.0×105g/mol);聚合动力学曲线表明,在60 min内随时间的延长,相对于催化剂17b,催化剂21的活性降幅更小。
3 [N,O]配体催化剂
1998年,Younkin等[36-37]研究了水杨醛亚胺中性镍配合物22(见图8)催化乙烯聚合的情况。以Ni(COD)2或B(C6F5)3为助催化剂时,该配合物可实现乙烯聚合。在5-位上引入强吸电子基团,催化剂的活性提高,产物支化度降低,且相对分子质量增大。水杨醛苯环的3-位上引入大位阻取代基可稳定催化剂的活性中心,起到屏蔽活性中心轴向的作用,催化活性提高,聚合物的相对分子质量和支化度也增大。
观察苯胺2,6-位芳基取代催化体系23(见图8)吸收乙烯的情况时发现,在60 ℃、1.0 MPa乙烯压力下,催化剂23a,23d,23e的活性能保持几个小时,而催化剂23b和23c在20 min内已完全失活[38]。这可能是由于苯环间位上的H原子与金属中心发生C—H键活化所致。
相对于催化剂24c(见图8),催化剂24a(见图8)具有更好的热稳定性[39]。在B(C6F5)3助催化作用下,当聚合温度由33 ℃升至63 ℃时,催化剂24a始终保持高活性。催化剂24c随聚合时间的延长活性降低,而催化剂24a在40 min内始终能保持较高活性。1H NMR表征结果显示,催化剂24a由于苯基提供了空间位阻,因此避免了因形成双配合物而造成的失活[40]。

图8 水杨醛亚胺等[N,O]配体镍催化剂Fig.8 Salicylaldimine and other[N,O] ligand nickel(Ⅱ) complexes.
Schrolder等[41]研究了一类由偶氮苯酚形成的中性镍配合物25(见图8),由于芳胺侧链上取代基的空间位阻减小,导致所得聚合产物的相对分子质量显著减小,即从聚合物变为低聚物。2-苯胺基环庚三烯酮的镍配合物26(见图8)可在80 ℃、不同乙烯压力下实现乙烯聚合,得到不同支化度的聚合产物,但此温度下催化剂的寿命短[42]。将羰基邻位用苯基或萘基取代,在40 ℃时催化剂寿命显著延长,半衰期大于1 h,且活性得以提高。
催化剂27a(见图8)经(Ni(COD)2或B(C6F5)3)活化后,在60 ℃、1.38 MPa乙烯压力下的活性很高,TOF=5×105h-1,但稳定性差,半衰期小于20 min。在35 ℃时催化剂27a的稳定性提高,半衰期超过15 h,且TOF达到106h-1[43]。若将苯胺2,6-位用大位阻基团取代,得到的催化剂27b可高活性地催化乙烯聚合,在70 ℃下反应1 h后,催化剂27b仍能大量吸收乙烯[44]。用[Ni(COD)2]活化催化剂27b后,乙烯快速吸收;体系反应温度由70 ℃升至100 ℃,聚合1 h后,乙烯吸收仍很强,表明催化剂27b在此温度下稳定性很好。
4 其他配体
在H(OEt2)2BAr4(Ar=3,5-(CF3)2C6H3,Et为乙基)的助催化作用下,催化剂28(见图9)催化乙烯聚合的热稳定性很好,甚至在100 ℃下该催化剂也没有分解,产物相对分子质量无下降趋势[45]。这种不同于α-二亚胺类似物的热稳定性可能来自于P原子对金属中心钯的良好配位能力。
用P原子取代α-二亚胺一边上的N原子,合成[P,N]配合物29(见图9)[46]。P比N具有更强的电子给予能力,更易与金属中心配位,从而提高配合物的热稳定性。经典α-二亚胺镍催化剂在70 ℃下的乙烯聚合反应中,往往在0.5 h内就分解,而催化剂29在70 ℃时可催化乙烯聚合长达几个小时,甚至在110 ℃下也可催化乙烯聚合,活性比70 ℃时高,但产物呈双峰分布,这可能是由于轴向的位阻不够大,发生了链转移,使产物的相对分子质量偏低。

图9 含[P,P][N,P]配体的催化剂Fig.9 Catalysts bearing[P,P][N,P] ligands.
5 结语
后过渡金属催化剂因具有优良特性而得到广泛研究,改进它的稳定性已逐渐被人们所重视。提高后过渡金属催化剂的稳定性可从3个方面考虑:1)适当提高配合物的空间位阻,以抑制C—N键的自由旋转,防止苯胺邻位上取代基与金属中心共平面而相互作用,降低C—H键活化所造成的催化剂失活;2)为金属中心提供合适的电子密度,配体所提供的适宜电子给予能力可稳定缺电子的14e-过渡态,抑制β-H消除,进而抑制链转移和催化剂失活;3)配体类型,不同类型的配体稳定性不同,不同配位原子对金属中心的不同配位能力直接影响催化剂的活性和稳定性。
提高后过渡金属催化剂的稳定性还有待进一步深入系统地研究,通过设计配合物以达到后过渡金属催化剂在高温下能长时间保持良好的催化性能,推动其工业化发展,促进它的实际应用。
[1] Peuckert M,Keim W. A New Nickel Complex for the Oligomerization of Ethylene[J]. Organometallics,1983,2(5):594 - 597.
[2] Camacho D H,Guan Zhibin. Designing Late-Transition Metal Catalysts for Olefin Insertion Polymerization and Copolymerization[J]. Chem Commun,2010,46(42):7879 - 7893.
[3] Johnson L K,Killian C M,Brookhart M. New Pd(Ⅱ)- and Ni(Ⅱ)-Based Catalysts for Polymerization of Ethylene and α-Olefins[J]. J Am Chem Soc,1995,117(23):6414 - 6415.
[4] Guan Zhibin,Cotts P M,McCord E F,et al. Chain Walking:A New Strategy to Control Polymer Topology[J]. Science,1999,283(5410):2059 - 2062.
[5] Ittel S D,Johnson L K,Brookhart M. Late-Metal Catalysts for Ethylene Homo- and Copolymerization[J]. Chem Rev,2000,100(4):1169 - 1203.
[6] Johnson L K,Mecking S,Brookhart M. Copolymerization of Ethylene and Propylene with Functionalized Vinyl Monomers by Palladium(Ⅱ) Catalysts[J]. J Am Chem Soc,1996,118(1):267 - 268.
[7] Gates D P,Svejda S A,Onate E,et al. Synthesis of Branched Polyethylene Using(α-Diimine) Nickel(Ⅱ) Catalysts:Influence of Temperature,Ethylene Pressure,and Ligand Structure on Polymer Properties[J]. Macromolecules,2000,33(7):2320 - 2334.
[8] Tempel D J,Johnson L K,Huff R L,et al. Mechanistic Studies of Pd(Ⅱ)-α-Diimine-Catalyzed Olefin Polymerizations[J].J Am Chem Soc,2000,122(28):6686 - 6700.
[9] Xie T,McAuley K B,Hsu J C C,et al. Gas Phase Ethylene Polymerization:Production Processes,Polymer Properties,and Reactor Modeling[J]. Ind Eng Chem Res,1994,33(3):449 - 479.
[10] Gottfried A C,Brookhart M. Living Polymerization of Ethylene Using Pd(Ⅱ) α-Diimine Catalysts[J]. Macromolecules,2001,34(5):1140 - 1142.
[11] Killian C M,Tempel D J,Johnson L K,et al. Living Polymerization of α-Olefins Using Ni(Ⅱ)-α-Diimine Catalysts:Synthesis of New Block Polymers Based on α-Olefins[J]. J Am Chem Soc,1996,118(46):11664 - 11665.
[12] Schmid M,Eberhardt R,Klinga M,et al. New C2v- and Chiral C2-Symmetric Olefin Polymerization Catalysts Based on Nickel(Ⅱ) and Palladium(Ⅱ) Diimine Complexes Bearing 2,6-Diphenyl Aniline Moieties:Synthesis,Structural Characterization,and First Insight into Polymerization Properties[J].Organometallics,2001,20(11):2321 - 2330.
[13] Meinhard D,Wegner M,Kipiani G,et al. New Nickel(Ⅱ)Diimine Complexes and the Control of Polyethylene Microstructure by Catalyst Design[J]. J Am Chem Soc,2007,129(29):9182 - 9191.
[14] Ionkin A S,Marshall W J. ortho-5-Methylfuran- and Benzofuran-Substituted η3-Allyl (α-Diimine) Nickel(Ⅱ) Complexes:Syntheses,Structural Characterization,and the First Polymerization Results[J]. Organometallics,2004,23(13):3276 - 3283.
[15] Camacho D H,Salo E V,Ziller J W,et al. Cyclophane-Based Highly Active Late-Transition- Metal Catalysts for Ethylene Polymerization[J]. Angew Chem Int Ed,2004,43(14):1821 - 1825.
[16] Popeney C S,Levins C M,Guan Zhibin. Systematic Investigation of Ligand Substitution Effects in Cyclophane-Based Nickel(Ⅱ) and Palladium(Ⅱ) Olefin Polymerization Catalysts[J]. Organometallics,2011,30(8):2432 - 2452.
[17] Camacho D H,Salo E V,Guan Zhibin,et al. Nickel(Ⅱ) and Palladium(Ⅱ) Complexes with an Alkane-Bridged Macrocyclic Ligand:Synthesis,Characterization,and Polymerization Test[J]. Organometallics,2005,24(21):4933 - 4939.
[18] Gates D P,Svejda S A,Onate E,et al. Synthesis of Branched Polyethylene Using (α-Diimine) Nickel(Ⅱ) Catalysts:Influence of Temperature,Ethylene Pressure,and Ligand Structure on Polymer Properties[J]. Macromolecules,2000,33(7):2320 - 2334.
[19] Liu Fengshou,Hu Haibin,Xu Ying,et al. Thermostable α-Diimine Nickel(Ⅱ) Catalyst for Ethylene Polymerization:Effects of the Substituted Backbone Structure on Catalytic Properties and Branching Structure of Polyethylene[J].Macromolecules,2009,42(20):7789 - 7796.
[20] Liu Jingyu,Li Yanguo,Li Yuesheng,et al. Ethylene Polymerization by (α-Diimine) Nickel(Ⅱ) Complexes Bearing Different Substituents on para-Position of Imines Activated with MMAO[J]. J Appl Polym Sci,2008,109(2):700 - 707.
[21] Popeney C S,Guan Zhibin. Effect of Ligand Electronics on the Stability and Chain Transfer Rates of Substituted Pd(Ⅱ)α-Diimine Catalysts[J]. Macromolecules,2010,43(9):4091 - 4097.
[22] Popeney C S,Levins C M,Guan Zhibin. Systematic Investigation of Ligand Substitution Effects in Cyclophane-Based Nickel(Ⅱ) and Palladium(Ⅱ) Olefin Polymerization Catalysts[J]. Organometallics,2011,30(8):2432 - 2452.
[23] Popeney C S,Rheingold A L,Guan Zhibin. Nickel(Ⅱ) and Palladium(Ⅱ) Polymerization Catalysts Bearing a Fluorinated Cyclophane Ligand:Stabilization of the Reactive Intermediate[J]. Organometallics,2009,28(15):4452 - 4463.
[24] Leung D H,Ziller J W,Guan Zhibin. Axial Donating Ligands:A New Strategy for Late Transition Metal Olefin Polymerization Catalysis[J]. J Am Chem Soc,2008,130(24):7538 - 7539.
[25] Huang Zengfang,Song Keming,Liu Fengshou,et al. Synthesis and Characterization of a Series of 2-Aminopyridine Nickel(Ⅱ) Complexes and Their Catalytic Properties Toward Ethylene Polymerization[J]. J Polym Sci,Part A:Polymer Chem,2008,46(5):1618 - 1628.
[26] Zai Shaobo,Liu Fengshou,Gao Haiyang,et al. Longstanding Living Polymerization of Ethylene:Substituent Effect on Bridging Carbon of 2-Pyridinemethanamine Nickel Catalysts[J]. Chem Commun,2010,46(42):4321 - 4323.
[27] Lee Jen-Jeh,Yang Fengzhao,Lin Yafan,et al. Unsymmetrical Bidentate Ligands of α-Aminoaldimines Leading to Sterically Controlled Selectivity of Geometrical Isomerism in Square Planar Coordination[J]. Dalton Trans,2008,43:5945 - 5956.
[28] Yang Fengzhao,Chen Yichun,Lin Yafan,et al. Nickel Catalysts Bearing Bidentate α-Aminoaldimines for Ethylene Polymerization-Independent and Cooperative Structure/Reactivity Relationship Resulting from Unsymmetric Square Planar Coordination[J]. Dalton Trans,2009,7:1243 - 1250.
[29] Gao Haiyang,Hu Haibin,Zhu Fangming,et al. A Thermally Robust Amine-Imine Nickel Catalyst Precursor for Living Polymerization of Ethylene Above Room Temperature[J].Chem Commun,2012,48(27):3312 - 3314.
[30] Small B L,Brookhart M,Bennett A M A. Highly Active Iron and Cobalt Catalysts for the Polymerization of Ethylene[J]. J Am Chem Soc ,1998,120(16):4049 - 4050.
[31] Britovsek G J P,Gibson V C,Kimberley B S,et al. Novel Olefin Polymerization Catalysts Based on Iron and Cobalt[J].Chem Commun,1998,7:849 - 850.
[32] Guo Lihua,Gao Haiyang,Zhang Ling,et al. An Unsymmetrical Iron(Ⅱ) Bis(imino)pyridyl Catalyst for Ethylene Polymerization:Effect of a Bulky Ortho Substituent on the Thermostability and Molecular Weight of Polyethylene[J].Organometallics,2010,29(9):2118 - 2125.
[33] Yu Jiangang,Liu Hao,Zhang Wenjuan,et al. Access to Highly Active and Thermally Stable Iron Procatalysts Using Bulky 2-[1-(2,6-Dibenzhydryl-4-Methylphenylimino)Ethyl]-6-[1-(Arylimino) Ethyl]Pyridine Ligands[J]. Chem Commun,2011,47(11):3257 - 3259.
[34] Wang Lincai,Sun Junquan. Methylene Bridged Binuclear Bis(Imino) Pyridyl Iron(Ⅱ) Complexes and Their Use as Catalysts Together with Al(i-Bu)3for Ethylene Polymerization[J].Inorganica Chimica Acta,2008,361(7):1843 - 1849.
[35] Liu Jingyu,Li Yuesheng,Liu Jingyao,et al. Ethylene Polymerization with a Highly Active and Long-Lifetime Macrocycle Trinuclear 2,6-Bis(Imino)Pyridyliron[J]. Macromolecules,2005,38(7):2559 - 2563.
[36] Younkin T R,Connor E F,Henderson J I,et al. Neutral,Single-Component Nickel(Ⅱ) Polyolefin Catalysts That Tolerate Heteroatoms[J]. Science,2000,287(5452):460 - 462.
[37] Wang Chunming,Friedrich S,Younkin T R,et al. Neutral Nickel(Ⅱ)-Based Catalysts for Ethylene Polymerization[J].Organometallics,1998,17(15):3149 - 3151.
[38] Zuideveld M A,Wehrmann P,Rohr C,et al. Remote Substituents Controlling Catalytic Polymerization by Very Active and Robust Neutral Nickel(Ⅱ) Complexes[J]. Angew Chem,Int Ed,2004,116(7):869 - 869.
[39] Song Dongpo,Wang Yongxia,Mu Hongliang,et al. Observations and Mechanistic Insights on Unusual Stability of Neutral Nickel Complexes with a Sterically Crowded Metal Center[J]. Organometallics,2011,30(5):925 - 934.
[40] Connor E F,Younkin T R,Henderson J I,et al. Synthesis of Neutral Nickel Catalysts for Ethylene Polymerization:The Influence of Ligand Size on Catalyst Stability[J]. Chem Commun,2003,18:2272 - 2273.
[41] Schrolder D L,Keim W,Zuideveld M A,et al. Ethylene Polymerization by Novel,Easily Accessible Catalysts Based on Nickel(Ⅱ) Diazene Complexes[J]. Macromolecules,2002,35(16):6071 - 6073.
[42] Hicks F A,Jenkins J C,Brookhart M. Synthesis and Ethylene Polymerization Activity of a Series of 2-Anilinotropone-Based Neutral Nickel(Ⅱ) Catalysts[J]. Organometallics,2003,22(17):3533 - 3545.
[43] Zhang Lei,Brookhart M,White P S. Synthesis,Characterization, and Ethylene Polymerization Activities of Neutral Nickel(Ⅱ) Complexes Derived from Anilino-Substituted Enone Ligands Bearing Trifluoromethyl and Trifluoroacetyl Substituents[J]. Organometallics,2006,25(8):1868 - 1874.
[44] Yu S M,Berkefeld A,Gottker-Schnetmann I. Synthesis of Aqueous Polyethylene Dispersions with Electron-Deficient Neutral Nickel(Ⅱ) Catalysts with Enolatoimine Ligands[J].Macromolecules,2007,40(3):421 - 428.
[45] Ikeda S,Ohhata F,Miyoshi M,et al. Synthesis and Reaction of Palladium and Platinum Complexes Bearing Diphosphinidenecyclobutene Ligand:A Thermally Stable Catalyst for Ethylene Polymerization[J]. Angew Chem,Int Ed,2000,39(24):4512 - 4513.
[46] Guan Zhibin,Marshall W J. Synthesis of New Phosphine Imine Ligands and Their Effects on the Thermal Stability of Late-Transition-Metal Olefin Polymerization Catalysts[J].Organometallics,2002,21(17):3580 - 3586.
