海藻酸钠对改性黏土絮凝特征的影响
2013-12-01林勇新曹西华宋秀贤俞志明中国科学院海洋研究所海洋生态与环境科学重点实验室山东青岛26607中国科学院大学北京00039
林勇新 ,曹西华 ,宋秀贤 ,俞志明 * (.中国科学院海洋研究所海洋生态与环境科学重点实验室,山东 青岛26607;2.中国科学院大学,北京 00039)
改性黏土治理有害藻华被认为是最具有发展前景的方法之一[1-2],具有经济、环保、高效[3-4]等优点.传统上对改性黏土絮凝微藻的研究,是把改性黏土颗粒及絮凝体假设为球形实心体,仅考虑了絮凝的宏观过程,并未考虑微藻的形态及其分泌的藻源有机质对于絮凝过程的影响.而大量研究业已证明,颗粒形态及水体中的有机大分子对于颗粒的絮凝过程有重要影响.Pivokonsky等[5]研究表明低浓度胞外有机质(EOM)通过表面吸附或架桥作用而提高水处理效率,而高浓度EOM因为增加了颗粒表面的负电荷而抑制絮凝过程;Guenther等[6]发现藻细胞的形态特征,如藻细胞大小、形状、表面积-体积比会影响黏土-藻细胞的絮凝过程;Verspagen等[7]研究发现藻细胞外糖醛酸的含量影响黏土对于藻细胞的絮凝效率.
分形理论是以自然界和非线性系统中出现的不光滑和不规则的几何形体为主要研究对象,认为分形是一类无规则、混乱而复杂,但其局部与整体有相似性的体系[8-9].分形理论在絮凝形态学研究中的应用解决了对絮凝过程中形态多变、结构复杂的絮凝体的定量描述问题.在絮凝过程中,分形维数会随着絮凝体形成条件的变化而变化,能够很好地描述和分析絮凝体的形成和生长机制.
海藻酸钠是从藻类和细菌中提取的一种高分子多糖化合物,是水体中天然有机质的重要组成成分[10-11].基于天然有机质种类繁多、成分复杂、不确定性强等特点,单一成分的海藻酸钠成为研究天然有机质在改性黏土颗粒上吸附特征及影响无机颗粒物对滤膜污染的理想模拟化合物之一[12-13].Jermann等[12]研究无机颗粒吸附天然有机物后,两者形成的结合物在超滤过程中产生的滤膜污染问题,就采用了海藻酸钠、腐植酸模拟了天然有机物在高岭土上的吸附特征.
本研究在前期研究基础上,以海藻酸钠为模式化合物,考察了藻源有机质对改性黏土絮凝速率的影响,分析了不同浓度海藻酸钠溶液中改性黏土颗粒物的絮凝动力学变化特征;并借助图像分析法,测定了该絮凝体的分形维数,利用分形维数对絮凝体的形态学特征进行了半定量化分析;探讨了藻源有机质对改性黏土絮凝体结构的影响.
1 材料与方法
1.1 材料与试剂
黏土矿物取自江苏苏州,主成分为高岭石68%,蒙脱石 25%,伊利石 5%,表面电位为-15.9mV,BET比表面积为44m2/g[14],使用前经表面改性处理[15-16].
海藻酸钠纯化:称量海藻酸钠(化学纯,国药集团化学试剂有限公司,黏度((10g/L,20)/℃(Pa·s)≥0.02)2.0g 于 50mL 蒸馏水中溶解,加入10% HCl溶液至pH2.0,边加边搅拌,逐渐出现海藻酸凝胶块;过滤后用蒸馏水洗两次,以除去附着在凝胶上多余的酸、无机盐、色素等;往海藻酸凝胶中加入1% Na2CO3溶液,调节pH7.0,充分搅拌,放置片刻,使海藻酸完全转化为海藻酸钠.海藻酸钠溶液中加入无水乙醇,边加边搅拌,海藻酸钠絮状物析出,过滤后放入烘箱,40℃下干燥,粉碎,得到海藻酸钠精制品.
100mg/L及1000mg/L海藻酸钠储备液的配备:准确称取0.1000g,1.000g海藻酸钠纯品置于1L烧杯中,加入少量二次蒸馏水,置于 55℃恒温水浴锅中加热,并用玻璃棒搅拌助溶待海藻酸钠全部溶解后转移至1L容量瓶中,冷却后用二蒸水稀释至1L,转移至试剂瓶中,4℃冰箱中保存备用.
1.2 絮凝动力学实验
根据实验设计,设置低浓度组和高浓度组,低浓度组的海藻酸钠浓度分别为 1.0,2.0,5.0,10.0,20.0mg/L;高浓度组分别为 50.0,100.0,500.0mg/L;取一定体积、不同梯度浓度的海藻酸钠溶液于50mL比色管中,设置空白组对照,每组设3个平行样,加入改性黏土达0.5g/L,迅速摇匀后,在室温、420nm波长下用1cm比色池测定不同时间的透光率(T%).
1.3 絮凝体图像采集及分形维数的测定方法
絮凝实验步骤同1.2,在20℃下静置3h.然后,利用移液管吸出上清液,并将絮凝体小心转移至显微镜载物片中.利用Nikon H600L 光学显微镜观察,在100×条件下拍照,图像通过CCD采集后传输到计算机,进行图像分析.
计算分形维数的方法主要包括周长-面积关系(或表面积-体积关系)求分维法、盒计数法、sandbox法、面积-回转半径法、变换法、密度-密度相关函数法等;其中,盒计数法由于其简单、方便等特点得到广泛使用.本研究的分维测定采用imageJ软件中的盒计数法进行絮体分维的测定,主要原理为:假定分形图片(即图像中含有图形像素部分)可以被边长为 ε的二维小方格完全覆盖起来,令N(ε)代表所需小方格的最低数目,即计算所需的格子数N(ε).为了减少误差,使不同尺寸的小方格能覆盖相同大小的图形,如 256×256像素的图形的ε应当是1、1/2、1/4、1/8、1/16、1/32、1/64…,直至降到 1/256.将一系列的 N(ε)、ε数据作㏑ N(ε)~㏑ N(ε)的双对数图,如能得到一条直线,则能说明N(ε)、ε有如下关系:

直线的斜率D就是图形的分形维数.如果㏑N(ε)~㏑(1/ε)图上只有一部分是直线时,则此图形的自相似性(标度不变性)只存在于直线部分的测度范围内.
2 结果与讨论
2.1 不同浓度海藻酸钠溶液中改性黏土颗粒物的絮凝动力学研究
根据碰撞理论及俞志明等[15]的研究表明,絮凝体颗粒间的絮凝碰撞可用双分子反应来处理,透光率与体系在t时刻总颗粒数间的关系可表示为:

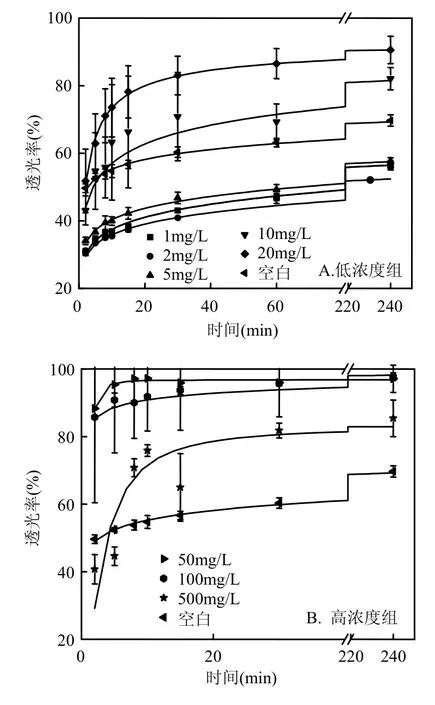
图1 不同浓度海藻酸钠对改性黏土絮凝速率的影响Fig.1 The effect of sodium alginate on the coagulation rate of modified clay
式中:T%为透光率;Nt为t时刻体系中粒子的总数;根据式(2)可得到透光率随时间的变化可以反映体系絮凝速率的大小.
图1是不同浓度海藻酸钠对改性黏土絮凝速率的影响,当海藻酸钠浓度低于 10mg/L 时,改性黏土的絮凝速率低于对照组,3 个浓度体系的絮凝速率差异不大;但在 10mg/L 时,絮凝效果已经高于对照组;随着海藻酸钠浓度增至 10~50mg/L,絮凝速率迅速提高且随着海藻酸钠浓度的增加而逐渐增大,在50mg/L时,达到最大值(表1).之后,当海藻酸钠浓度继续增大至 50~100mg/L,并不能继续促进改性黏土颗粒物间絮凝速率的提高,如50、100mg/L组的絮凝速率相同,且均能迅速(5min内)达到絮凝平衡,即ΔT%迅速降低至0;当海藻酸钠浓度大于100mg/L时,絮凝速率降低,但絮凝效果依然高于对照组.
海藻酸钠是具有高分子链的亲水性多糖化合物(图 2),在水溶液中容易电离产生带有-COO-、-OH-等亲水负电荷基团[17].俞志明等[16]研究表明经无机改性后的黏土表面的电性会发生反转,ζ电位由负值转变为正值,因此实验中黏土的颗粒均带正电荷.在低浓度(<10mg/L)海藻酸钠溶液的改性黏土体系中,由于海藻酸钠电离的负电荷基团通过电中和吸附在黏土表面,导致黏土颗粒的正电性减少,颗粒间的相互吸引作用能减小,有效碰撞次数降低[18],絮凝速率降低;当海藻酸钠浓度升至 10mg/L时,海藻酸钠大分子中的长碳链憎水结合作用增加,延长絮凝了时间,絮凝效果提高. 随着海藻酸钠浓度的增加(10~50mg/L),可能是由于大量吸附于改性黏土表面的海藻酸钠的高分子长链增加了改性黏土颗粒的有效作用半径,增加颗粒间的范德华力作用[19],从而提高絮凝速率和絮凝效果.然而这种由范德华力引起的吸附网捕作用比由化学反应作用力产生的吸附作用要弱的多,在海藻酸钠达到一定浓度(50~100mg/L)时,范德华力引起的较弱的网捕作用在絮凝体外围可能达到了一个平衡,即絮凝体增加颗粒层与脱离的颗粒达到平衡,絮凝速率也达到了一个平台.当海藻酸钠浓度继续增加时(达到 500mg/L),这种平衡被打破,同时大量海藻酸钠分子附着在改性黏土颗粒表面降低了有效碰撞效率,导致絮凝速率降低;但由于憎水结合作用,絮凝作用效果仍高于对照.

图2 海藻酸钠分子结构Fig.2 The molecular structure of sodium alginate
可见,当海藻酸钠浓度介于 10~100mg/L时,可以有效提高改性黏土絮凝的速率;而低于或高于这个浓度范围时,海藻酸钠都会降低改性黏土的絮凝速率.另一方面,实验结果也表明,当海藻酸钠浓度≥10mg/L后,适当延长絮凝时间,其对改性黏土的絮凝效果都有所提高.

表1 不同浓度的海藻酸钠与改性黏土的最大絮凝速率Table 1 Maximum coagulation rates of the modified clay at various concentration of sodium alginate
2.2 不同浓度海藻酸钠溶液中改性黏土颗粒物絮凝体分形维数的测定
2.2.1 测定分形维数方法的校正 采用盒子计数法对絮凝体的分形维数进行测定,为确保验证所使用的imageJ盒子计数计算分形维数的有效性.实验中对所使用的测定方法进行校正,表2为利用imageJ盒子计数法测定常规分形图像的分形维数,分别为Koch曲线1.2704,sierpinski三角形 1.5822,sierpinski正方形 1.8106,vicsek图形1.5027,与理论值的误差分别为 0.67%,0.18%,4.35%,2.57%,在合理范围内,证明了该分析方法的可靠性和准确性.
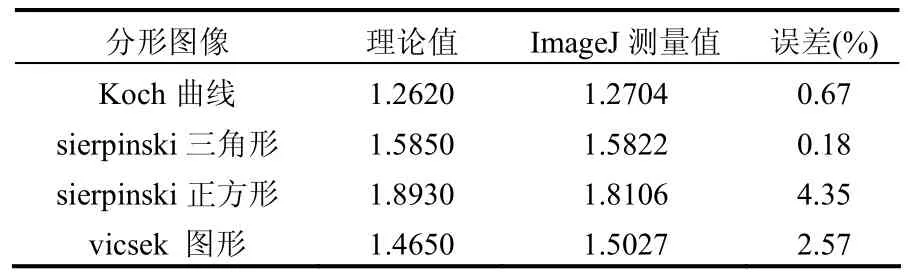
表2 ImageJ软件盒计数分维法对常规分形图像的计算值Table 2 Fractal dimension of standard fractal images by ImageJ fractal box count method
2.2.2 絮凝体分形维数的测定

图3 不同浓度海藻酸钠与改性黏土的絮凝体图像Fig.3 Images of clay flocculation with different concentrations of sodium alginate
通过对不同浓度海藻酸钠溶液中改性黏土颗粒物的絮凝体(图 3)分形维数的测定可知(表3),低浓度(<10mg/L)海藻酸钠溶液体系的絮凝体分形维数相应于对照组(Df=1.5246)略微降低,1,2,5mg/L组的分形维数分别为1.4838,1.4915,1.5121,但组间的变化不大;在低浓度的海藻酸钠溶液中,由于异电荷间的相互吸引作用,带有-COO-、-OH-等负电荷基团的海藻酸钠分子吸附在改性黏土的正电中心位置,这种正负电荷中心的强力结合作用以及海藻酸钠分子链上的羧基、羟基缔合形成的(如-C=O-OH)氢键作用[20]和改性黏土铝离子键(如-Al-OH)作用[21]使得吸附的海藻酸钠分子以平伏在改性黏土颗粒表面为主,如图 4A所示.这些海藻酸钠分子降低了改性黏土表面的正电性以及颗粒间的相互作用能,从而降低了改性黏土颗粒之间絮凝聚结的速率;此条件下形成的絮凝体由于颗粒之间作用能较低,分形维数要低于对照组[18].
随着海藻酸钠浓度增大,吸附在改性黏土颗粒表面的海藻酸钠分子逐渐增多,改性黏土颗粒表面正电性逐渐减少,导致改性黏土颗粒之间的静电作用逐渐减弱,海藻酸钠分子链逐渐由平伏转为矗立状,如图4B所示. 此时海藻酸钠大分子链的桥连作用逐渐增强,极大提高了改性黏土颗粒的作用半径[19]:由偶极-偶极相互作用形成的范德华力[22]的增强作用幅度要远大于改性黏土颗粒表面的静电作用的减弱幅度,综合两种作用改性黏土颗粒之间的聚结能力是逐渐增强的,因而所形成的絮凝体越加密实,分形维数逐渐增大;表现为分形维数在海藻酸钠浓度增大至10mg/L时迅速增加为 1.5893,并随着海藻酸钠浓度的增大而继续增大,如20mg/L为1.6388,到50mg/L时达到最大值 1.6823;说明海藻酸钠的高分子链在絮凝中的吸附架桥作用明显,絮凝体颗粒间的有效碰撞增大.

图4 海藻酸钠大分子吸附于改性黏土颗粒表面的模式Fig.4 The absorption diagram of sodium alginate on modified clay
此后,进一步加大海藻酸钠浓度,絮凝体的分形维数反而降低,100,500mg/L组分别为 1.5866,1.5639;其原因是当海藻酸钠分子在改性黏土上的吸附由有单分子层转为双层或多层时,海藻酸钠大分子链之间在改性黏土表面相互聚结(如图4C)其桥连功能降低;又由于海藻酸钠分子长碳链的憎水特征[23],大分子的负电中心外移[22],增大了改性黏土颗粒之间的静电排斥力.因此在此浓度范围内,海藻酸钠消弱了改性黏土颗粒间的聚结作用,形成絮凝体的空隙率逐渐增大,分形维数又逐渐降低[18];而絮凝体的破碎速率逐渐增大[24],即形成的絮凝体外围容易脱离絮凝体,从而表现为絮凝速率在浓度为500mg/L时再次降低.
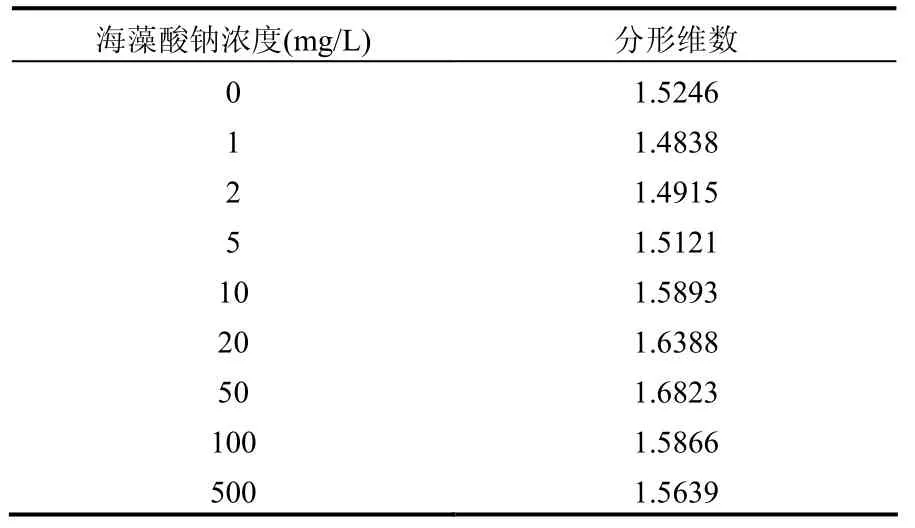
表3 ImageJ软件盒计数法对絮凝体分形图像的计算值Table 3 Fractal dimension of flocs by ImageJ fractal box count method
2.3 海藻酸钠对絮凝体形成的影响
Gregory[25]研究认为絮凝体的形成过程应该包括聚集的颗粒必须相互碰撞和依靠碰撞黏结在一起两个独立的步骤;因此,有效碰撞是决定絮凝体成长速率的关键因子.当絮凝体成长到一定状态时,絮凝体受剪切力、水的阻力等影响会发生破碎.在絮凝体成长过程中一般伴随着絮凝体的破碎,絮凝体的成长速率(Rfloc)由颗粒的碰撞速率(Rcol)和破碎速率(Rbr)共同决定[26],可表示为:

式中:α为有效碰撞系数,受到剪切力、絮凝体强度等多方面因素的影响;絮凝体强度是作为衡量絮凝体结构的一个重要参数,取决于颗粒间结合键的作用能大小以及数量,即絮凝体间的黏结力.
根据絮凝体的形成条件等[27-28],黏结力B,(g·cm)/s2与絮凝体的净截面积An(cm2)成正比:

式中:σ为常数,表示与絮凝体净截面积有关的平均黏结强度, g/(cm·s2).对一个特征长度为 d(cm)絮凝体净截面积An与絮凝体的分维二维Df的关系则有:

由此黏结力可表示为:

在本实验中,不同浓度海藻酸钠溶液在添加改性黏土后,在同样的搅拌条件下混匀后,静置.因此,在不考虑剪切力等影响因素的条件下,黏结力是影响絮凝体成长的主要影响因子.在低浓度(≤5mg/L)海藻酸钠溶液中,海藻酸钠在改性黏土上的吸附还不足于改变黏土颗粒的半径 d,因此,絮凝体颗粒的半径的变化可忽略不计;而此时形成的絮凝体的分形维数 Df低于对照组(表 3),由式(6)可知,在低浓度(<10mg/L)海藻酸钠溶液中絮凝体的黏结力低于对照组,絮凝体的强度减弱,改性黏土颗粒间有效碰撞次数降低;随海藻酸钠浓度的增大,组间絮凝体的分形维数变化不大,所以低浓度组(<10mg/L)组间的差异不明显.随着海藻酸钠浓度增加(10~50mg/L),大量海藻酸钠分子吸附在黏土颗粒上,增大了颗粒的有效作用半径;在此浓度范围内,由于絮凝体的特征半径及其分形维数都随着海藻酸钠浓度的增大而增大,所以絮凝体的黏结力迅速增加,絮凝体的絮凝强度迅速增大,提高了絮凝体的成长速率;并在50mg/L时,在单位时间内,成长速率率 Rfloc达到最大值.当海藻酸钠浓度大于 50mg/L之后,絮凝体的半径增大,但是絮凝体的分形维数降低;说明絮凝体的结构较为松散,空隙率较大,易发生破碎[24].由式(3)可知,当 αRcol=Rbr时,絮凝体的成长速率为 0,絮凝体的成长体系达到一个平衡.当海藻酸钠浓度为100mg/L时,改性黏土的絮凝速率与50mg/L浓度组相当(图1),说明在此浓度范围内,絮凝体的成长体系能在短时间内就达到一个平衡的状态.当海藻酸钠浓度继续增大(>100mg/L)时,絮凝体的空隙率、破碎率增大,分形维数及有效碰撞效率降低;此时 αRcol<Rbr,絮凝体的成长率 Rfloc为负值,其表现为图 1中500mg/L组达到絮凝平衡时间较其他组长,这也说明絮凝体的空隙率较大并促进了絮凝体的破碎,从而延长了絮凝的平衡时间.
可知,海藻酸钠在10~50mg/L浓度范围内可以提高改性黏土颗粒的黏结力及改性黏土颗粒的絮凝强度,从而增加改性黏土颗粒的有效碰撞次数、提高黏土颗粒的絮凝速率.
3 结论
3.1 溶液中的海藻酸钠对改性黏土絮凝有显著影响.10~100mg/L的海藻酸钠可以提高改性黏土颗粒物絮凝的速率,最佳浓度为 50mg/L;在浓度大于10mg/L而低于50mg/L时,改性黏土的絮凝速率、絮凝体的分形维数和絮凝体强度均随着海藻酸钠浓度的增大而增大,形成的絮凝体结构较为密实;浓度高于50mg/L而低于100mg/L时,改性黏土颗粒形成的絮凝体结构较为松散,分形维数降低、絮凝体强度减小.
3.2 低浓度(<10mg/L)海藻酸钠不利于改性黏土颗粒间的絮凝.高浓度(>100 mg/L)海藻酸钠能促进改性黏土的絮凝效率,但促进作用呈现减小的趋势,改性黏土颗粒形成的絮凝体结构松散、空隙率增大,分形维数降低.
[1]Anderson D M. Approaches to monitoring, control and management of harmful algal blooms (HABs)[J]. Ocean and Coastal Management, 2009,52:342-347.
[2]Kim H G. Mitigation and controls of HABs [M]//Ecology of harmful algae. Volume 189. New York: Springer Berlin Heidelberg, 2006:327-338.
[3]吴 萍,俞志明.新型黏土改性剂-烷基多糖苷季铵盐 [J]. 中国环境科学, 2006,26(6):680-684.
[4]李明玉,潘 倩,王丽燕,等.不同混凝剂对流溪河水源水中藻类去除对比 [J]. 中国环境科学, 2010,30(11):1484-1489.
[5]Pivokonsky M, Kloucek O, Pivokonska L. Evaluation of the production, composition and aluminum and iron complexation of algogenic organic matter [J]. Water Research, 2006, 3045-3052.
[6]Guenther M, Bozelli R. Factors influencing algae–clay aggregation [J]. Hydrobiologia, 2004,523:217-223.
[7]Verspagen J M H., Visser P M, Huisman J. Aggregation with clay causes sedimentation of the buoyant cyanobacteria Microcystis spp. Aquatic Microbial Ecology, 2006,44:165-174.
[8]张济忠.分形 [M]. 北京: 清华大学出版社, 1995.
[9]王毅力,李大鹏,解明曙.絮凝形态学研究及进展 [J]. 环境污染治理技术与设备, 2003,4(10):1-9.
[10]Fourest E, Volesky B. Alginate properties and heavy metal biosorption by marine algae [J]. Applied Biochemistry and Biotechnology, 1997,67:215-226
[11]Boyen C, Kloareg B, Polne-Fuller M, et al. Preparation of alginate lyases from marine molluscs for protoplast isolation in brown algae [J]. Phycologia, 1990,29:173-181.
[12]Jermann D, Ponk W, BOLLER M. Mutual Influences between Natural Organic Matter and Inorganic Particles and Their Combined Effect on Ultrafiltration Membrane Fouling [J].Environmental Science and Technology, 2008,42:9129-9136.
[13]Higgin R, Howe K J, Mayer T M. Synergistic behavior between silica and alginate: Novel approach for removing silica scale from RO membrances [J]. Desalination, 2010,250:76-81.
[14]王洪亮.颗粒物对藻华生物的絮凝作用及其分形数值模拟研究[D]. 青岛:中国科学院海洋研究所, 2010:25-35.
[15]俞志明,邹景忠,马锡年.粘土矿物去除赤潮生物的动力学研究[J]. 海洋与湖沼, 1995,26(2):1-6.
[16]俞志明,邹景忠,马锡年. 一种提高粘土矿物去除赤潮生物能力的新方法 [J]. 海洋与湖沼, 1994,25(2):226-232.
[17]Draget K I, Skjak-Braek G, Smidsrod O. Alginic acid gels: the effect of alginate chemical composition and molecular weight [J].Carbohydrate Polymers, 1994,25:31–38.
[18]Lin M Y, Lindsay H M, Weitz D A, et al. Universality in colloid aggregation [J]. Nature, 1989,339:360-362.
[19]Zhou Y, Franks G V. Flocculation mechanism induced by cationic polymer investigated by light scattering [J]. Langmuir,2006, 22:6775-6784.
[20]Liang C X, Hirabayashi K. Improvements of the physical properties of fibroin membranes with sodium alginate [J]. Journal of applied polymer science, 1992,45:1937-1943.
[21]Jackson M L. Aluminum bonding in soils: a unifying principle in soil science [J]. Soil Science Society of America Journal, 1962,27: 1-10.
[22]Yariv A, Yeh P. Optical waves in crystals [M]. New York : John Willey and Sons, 1984.
[23]Kang H A, Jeon G J, Lee M Y, et al. Effectiveness test of alginate-derived polymeric surfactants [J]. Journal of Chemical Technology and Biotechnology, 2002,77:205-210.
[24]李冬梅,谭万春,黄明珠,等.絮凝体的分形特性研究[J].给水排水,2004,30(5):5-9.
[25]Gregory J. The role of floc density in solid-liquid separation [J].Filtration and Separation.1998,35:367-371.
[26]Jarvis P, Jefferson B, Gregory J, et al. A review of floc strength and breakage [J]. Water Research, 2005,39:3121-3137.
[27]Tambo N. Physical aspect of flocculation process-Ⅰ:Fundamental treatise [J]. Water Research, 1979,13(5):429-439.
[28]金鹏康,曹 宇,王晓昌.絮凝体的物理特性 [J]. 西安建筑科技大学学报(自然科学版), 2001,33(4):316-320.
