生物碱样品中有效成分的CZE法分离与测定
2013-11-28高苏亚王树春
高苏亚,王 黎,王树春,李 华
(1.西安医学院 药学院,陕西 西安 710021;2.西北大学 分析科学研究所,陕西 西安 710069)
咖啡因、茶碱、可可碱是被广泛用于食品和药品的甲基黄嘌呤类生物碱,因具有多种生理活性而倍受青睐[1]。但高剂量或长期服用易产生成瘾性,甚至会导致焦虑、头痛、失眠、心律不齐等不良反应,被国际奥林匹克协会(IOC)定位为违禁物质。由于咖啡因及黄嘌呤类化合物广泛存在于日常饮食中[2],IOC已规定尿液中咖啡因的检测浓度不得超过12 ng·L-1[3]。因此建立对这3种结构非常相似的生物碱在实际样品中的分离和测定方法尤为必要。已有文献报道用HPLC结合紫外、安培等检测器[4-5]、TLC[6]、离子色谱[7]和 CE[8-12]检测食物和药物制剂中的黄嘌呤类生物碱,其中 HPCE 以其分离高效[13]、自动化程度高、试剂消耗量少、快速方便等特点而更具优势。但文献报道中的CE法大多仅限于其中一种或两种生物碱有效成分的测定,或是某一种样品的测定。本文以β-环糊精(β-CD)为添加剂,采用CE中最为简单的区带毛细管电泳法(CZE)分离测定了茶叶、饮料类、咖啡类和药品制剂中3种黄嘌呤类有效成分,为3种生物碱在各类实际样品中的测定提供了参考。
1 实验部分
1.1 仪器与试剂
P/ACETMMDQ型高效毛细管电泳仪(美国Beckman公司),配有二极管阵列检测器(DAD)和32Karat Software色谱工作站;熔融未涂层石英毛细管柱(河北永年光导纤维厂);KQ5200DB型数控超声波清洗器(昆山市超声仪器有限公司);BSZ10S电子天平(德国Sartorius公司);PB-10型酸度计(德国Sartorius公司)。
咖啡因、茶碱、可可碱标准品均购自中国药品生物制品检定所。茶叶、可乐罐装饮料、雀巢咖啡购自西安市人人乐超市,复方茶碱片和复方茶碱麻黄碱片购自西安市友谊医院,实验用水均为二次蒸馏水。
1.2 溶液制备
标准溶液:精密称取一定量咖啡因、茶碱、可可碱对照品,分别用水配制成质量浓度均为7.20 g·L-1水溶液,于4℃冰箱保存,使用前混合并稀释至所需浓度。
供试品溶液:精密吸取10.0 mL可口可乐罐装饮料于25 mL容量瓶中,加水定容,摇匀;取约2 g茶叶或1 g雀巢咖啡,精密称定,置于100 mL烧瓶中,加入50 mL 70℃的水搅拌使其溶解,水煮30 min,冷却后过滤,反复两次,将滤液合并,精密吸取5.0 mL于50 mL容量瓶中,加水定容,摇匀待用;取20片复方茶碱片或复方茶碱麻黄碱片,精密称定,研细,取约0.5 g药品粉末,精密称定,置于烧瓶中加50 mL水超声(150 W,40 kHz)溶解30 min,精密移取5.0 mL于50 mL容量瓶中,加水定容,摇匀待用。上述溶液进样前均经0.45 μm微孔滤膜过滤。
1.3 毛细管电泳条件
采用未涂层石英毛细管(75 μm ×60 cm,有效长度50 cm),以20 mmol·L-1硼砂 -4 mmol·L-1β-CD(pH 9.0)为运行缓冲液,压力(0.5 psi)进样5 s,运行电压16 kV,检测波长273 nm,温度25℃。每次实验前分别用0.1 mol·L-1HCl、水、0.1 mol·L-1NaOH、水和缓冲液各冲洗毛细管10 min。每两次运行间分别用水和缓冲液各冲洗3 min。
2 结果与讨论
2.1 检测波长的选择
经紫外光谱扫描,咖啡因、茶碱、可可碱在273 nm附近均有最大吸收,因此选择273 nm作为本实验的检测波长。
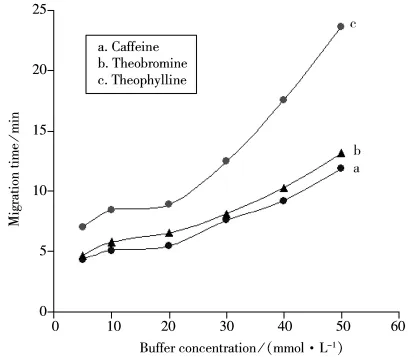
图1 缓冲液浓度对3种生物碱迁移时间的影响Fig.1 Effect of buffer concentration on migration time of three alkaloids
2.2 缓冲液浓度的选择
选用硼砂缓冲体系,基线较平稳。本实验考察了硼砂浓度为5.0~50.0 mmol·L-1时对3种生物碱分离的影响(见图1)。结果发现,硼砂浓度较低时,咖啡因和可可碱较难分离,随着硼砂浓度的增大其分离度有所提高,但分析时间大大延长,同时电流也随之升高(当缓冲液浓度为40 mmol·L-1时,电流已接近100 μA),同时茶碱的峰拖尾现象加重。综合考虑峰形、峰电流强度和分析时间,最终选择硼砂的最佳浓度为20 mmol·L-1。
为了进一步改善咖啡因与可可碱之间的分离度和峰形,实验考察了向硼砂溶液中加入不同浓度β-CD对分析的影响。结果发现,加入β-CD后3种生物碱的分离度得到显著改善,特别是咖啡因和可可碱明显可达到基线分离,三者的峰形也得到不同程度的改善。这可能是因为呈截顶圆锥状结构含7个葡萄糖单元的β-CD具有“内疏水、外亲水”的特征,与药物分子间通过非共价作用力相互作用,从而对待测物具有选择性匹配或选择性识别,使其分离度增大并改善峰形。但迁移时间随着β-CD浓度的增加而延长,当β-CD浓度较高时会使得焦耳热效应较为显著,从而导致基线不平和谱带展宽。因此,综合考虑后选择β-CD的最佳浓度为4 mmol·L-1。
2.3 缓冲液pH值的选择
缓冲液的pH值能够影响毛细管内壁硅羟基的解离,并影响待测物的表观电荷和电渗流,进而影响被测物的分离度。用硼酸将缓冲液pH值调至7.0~9.0,用NaOH将其pH值调至9.5~11.0,其他条件不变,对3种生物碱的混合标准品溶液进行分析。结果发现,当pH 7.0时,3种生物碱谱峰均有重叠,增大pH值时三者的分离度增大但同时迁移时间也不断增大,pH 9.0时三者可达到完全基线分离;继续增大缓冲液pH值时,分析时间明显延长,且电流明显上升,焦耳热增大,尤其是茶碱谱峰严重展宽。因此选择缓冲液的最佳pH值为9.0。
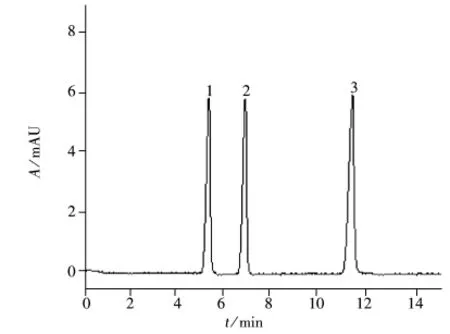
图2 优化条件下3种生物碱的电泳谱图Fig.2 Electrophoregram of three alkaloids at optimized conditions
2.4 运行电压的选择
考察了电压在5~25 kV范围内对3种生物碱分离的影响。结果发现,随着施加电压的增大,分析时间明显减少,峰强度也有所增加,但电压过高时分离度显著下降,峰电流明显增大,基线噪音变大,影响检测的灵敏度和稳定性。当电压为16 kV时,3种生物碱的分析时间、分离效果和峰电流均较为理想,因此选择运行电压为16 kV。
实验还考察了进样时间、毛细管柱温等因素对分析的影响。结果发现,进样时间为5 s(0.5 psi),温度25℃时,3种生物碱的分离度、分析时间、峰形和峰电流均较佳。
综上所述,确定最佳CZE条件见“1.3”。在此条件下3组分的谱图见图2。
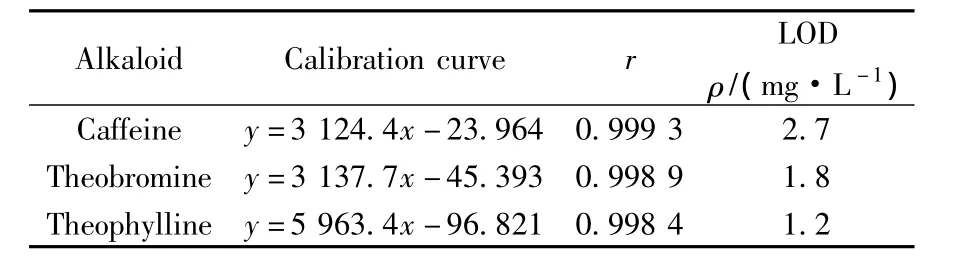
表1 3种生物碱的线性关系和检出限Table 1 Calibration relationship and detection limits(LODs)of alkaloids
2.5 线性范围与检出限
分别精密移取浓度均为0.72 g·L-1的混合标准溶液 0.5、1.0、1.5、2.0、2.5、3.0、3.5、4.0 mL于10 mL容量瓶中,定容。在上述条件下分别连续进样3次,以3种生物碱的峰面积(y)对其质量浓度(x)进行拟合,线性关系见表1。3种生物碱在0.036~0.288 g·L-1范围内线性关系良好,其检出限均不大于2.7 mg·L-1。
2.6 精密度、重现性与稳定性
将含咖啡因、茶碱和可可碱浓度均为0.144 g·L-1的标准混合溶液连续进样6次,测得3种生物碱峰面积的RSD均小于2.4%。取“1.2”的可口可乐罐装饮料供试品溶液连续进样6次,结果测得咖啡因峰面积的RSD为1.9%,随后分别间隔1、2、4、8、16、24 h进样,测得其峰面积的RSD为3.2%。说明样品在该实验条件下的重现性较好,在24 h内基本稳定。
2.7 样品加标回收实验
精密吸取3种对照品储备液适量(高、中、低含量),分别加入到已知含量的信阳产绿茶样品中,按“1.2”方法制备供试品溶液,测得3种生物碱的回收率见表2。由表2结果可知,3种生物碱的回收率为97%~104%,RSD均小于4.0%,满足毛细管电泳对样品定量的要求。
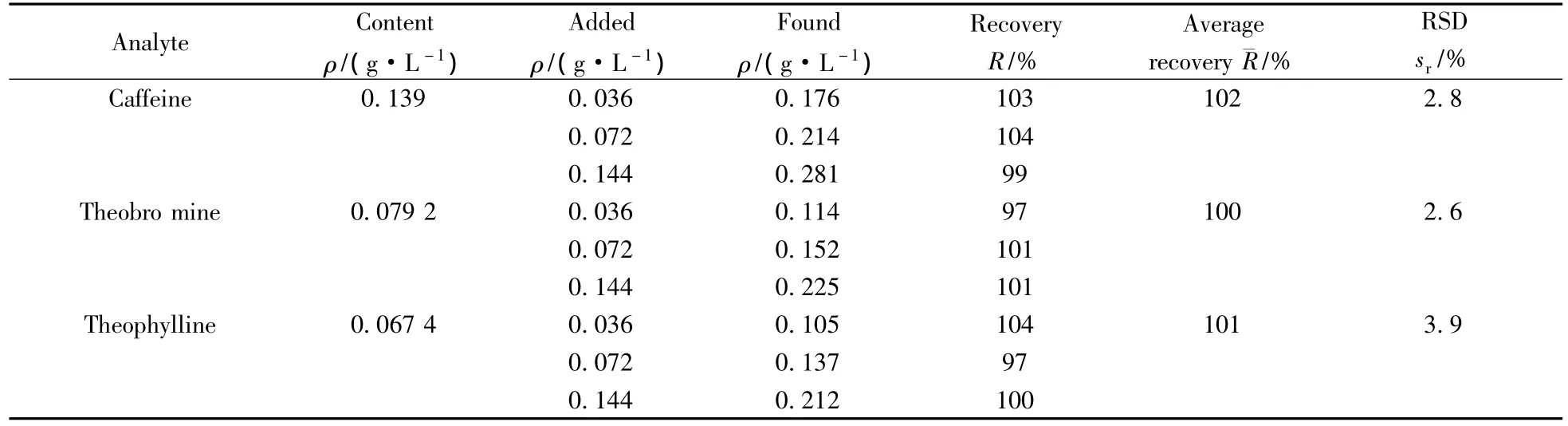
表2 加样回收率实验结果Table 2 Results of recovery test
2.8 实际样品测定
在上述优化实验条件下,实际样品中的3种生物碱可得到较好的分离与检测(见图3)。可乐饮料、茶叶、咖啡、药品等4类实际样品中3种生物碱的测定结果见表3。
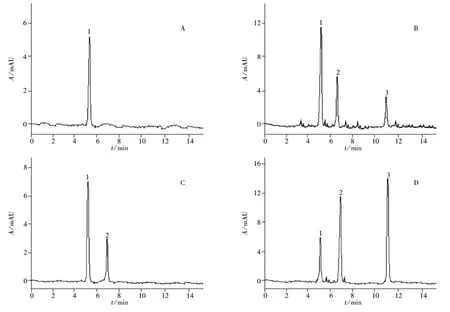
图3 实际样品的电泳谱图Fig.3 Electrophoregrams of some real samples
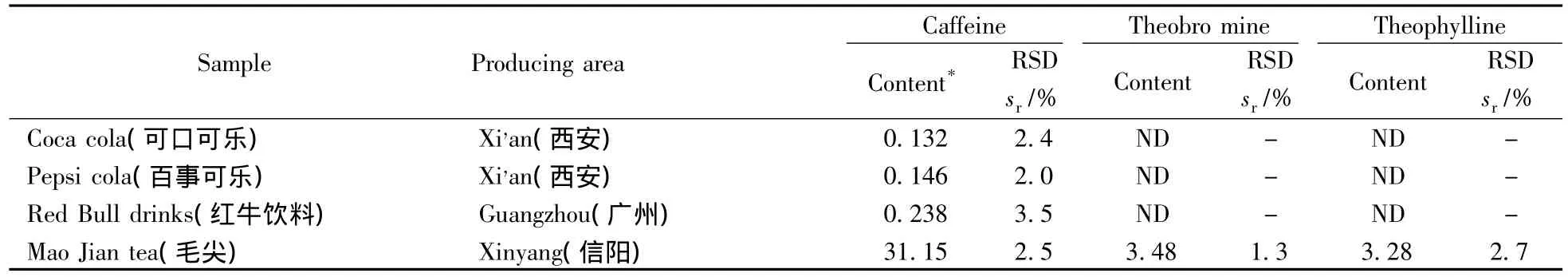
表3 实际样品中3种生物碱的含量测定结果Table 3 Determination results of three alkaloids in real samples
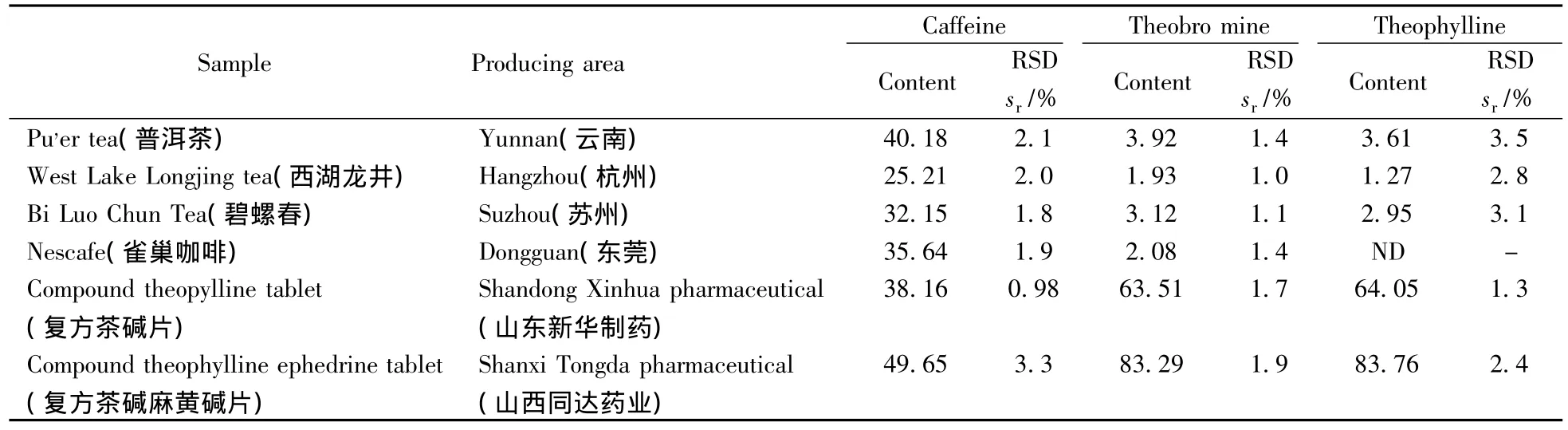
(续表3)
3 结论
本文建立了以β-CD为添加剂的CZE法对样品中咖啡因、可可碱、茶碱3种生物碱同时定量的分析方法。考察了硼砂缓冲液浓度、β-CD添加剂浓度、缓冲液pH值以及电泳电压等因素的影响,确定较优化的电泳条件为:20 mmol·L-1硼砂+4 mmol·L-1β-CD(pH 9.0)作为运行缓冲液,工作电压16 kV,进样时间5 s(0.5 psi),检测波长273 nm,温度25℃。在该实验条件下,3种生物碱的电泳谱图峰面积与其质量浓度在0.036~0.288 g·L-1范围内呈良好线性,r≥0.998 4;精密度、重复性和稳定性良好,其峰面积的RSD均不大于3.2%,其检出限均不大于2.7 mg·L-1,加标回收率为97%~104%,方法快速简便,灵敏度高,适用面广,可用于多种样品种类(包括饮料、茶叶、咖啡和药品等)的分析。
[1]McGaw L J,Steenkamp V,Eloff J N.J.Ethnopharmacol.,2007,110(1):16 -20.
[2]Gunasekaran S,Sankari G,Ponnusamy S.Spectrochim.Acta A,2005,61(1/2):117 -127.
[3]Pérez-Marinez I,Sagrado S,Medina- Hernández M J.Anal.Chim.Acta,1995,304(2):195 -201.
[4]Rodrigues C I,Marta L,Maia R,Miranda M,Ribeirinbo M,Maguas C.J.Food Compos.Anal.,2007,20(5):440 -448.
[5]Meyer A,Ngiruwonsanga T,Henze G.J.Anal.Chem.,1996,356(3/4):284 -287.
[6]Li B Y,Tong L,Zou B T.Acta Pharm.Sin.(李炳阳,童路,邹本田.药学学报),1985,20(5):398-400.
[7]Chen Q C,Wang J.J.Chromatogr.A,2001,937(1/2):57 -64.
[8]Cianchino V,Acosta G,Claudia O,Martinez L D,Gomez M R.Food Chem.,2008,108(3):1075-1081.
[9]Tagliaro F,Smith F P,Turrina S,Eauisetto V,Marigo M.J.Chromotagr.A,1996,735(1/2):227 -235.
[10]Injac R,Srdjenovic B,Prijatelj M,Boskovic M,Karljikovic-Rajic K,Strukelj B.J.Chromatogr.Sci.,2008,46(2):137-143.
[11]Li M J,Zhou J Y,Gu X,Wang Y,Huang X J,Yan C.J.Sep.Sci.,2009,32(2):267 -274.
[12]Zhu L,Shen G J,Wang L L.J.Anal.Sci.(祝玲,申贵隽,王莉莉.分析科学学报),2011,27(4):422-426.
[13]Li S M,Ji H,Huang B Y,Shen Q,Yuan M.J.Instrum.Anal.(李士敏,季红,黄碧云,申青,袁牧.分析测试学报),2010,29(4):376-378.
