蛋白质分子待定模板印迹聚合物的合成及应用
2013-11-28栾新杰赵美凤胡效亚
杨 春,栾新杰,赵美凤,胡效亚
(扬州大学 化学化工学院,江苏 扬州 225002)
分子印迹聚合物(MIPs)是在特殊模板分子存在下制备的材料[1],当除去模板分子后,该聚合物具有选择亲和性,能够吸引或识别原来的模板。MIPs在诸如传感器[2]、催化[3-5]、分离[6-8]、分子识别[9-11]、抗体模拟[12]以及晶体工程[13]等领域发挥重要作用。蛋白质印迹聚合物通常按照非共价方法合成[14-17]。低位阻的 MIP 由表面印迹[18-19]、纳米印迹[20]或冻胶[21]技术得到。这些材料能够保证模板与最终聚合物之间的快速吸附-脱附平衡。
复杂蛋白质分析,尤其是蛋白质组学研究中,降低样品的复杂性非常重要。到目前为止,很少有方法在脱除高丰度蛋白质的同时,能够富集样品中的低丰度组分。一种名为“固态配体库”(Solidphase ligand library)的方法,可从复杂样品中同时萃取高、低丰度蛋白质,具有降低主要成分、同时浓缩低丰度成分的作用[22]。但此法有一定局限性,必须根据样品性质设计、合成特殊的配体库。
本文研究了一种蛋白质印迹聚合物的新合成方法,既不使用标准蛋白质,也无需合成任何配体。在样品(鸡蛋清)存在下,基于冻胶方法合成聚合物。聚合物在毛细管中形成,从而得到整体柱。同时在相同条件下合成了一根对比柱。通过对比研究,发现样品中多个高丰度组分被印迹,印迹整体柱可从样品溶液中特异地吸附作为印迹模板的高丰度组分,同时增强低丰度蛋白质的信号。
1 实验部分
1.1 仪器与试剂
HPCE-10型毛细管电泳仪(大连江申分离科学仪器公司),紫外吸收检测器,采用电渗流进样;毛细管(内径100 μm,外径365 μm)购自河北省永年锐沣色谱器件有限公司;缓冲溶液为10 mmol/L(pH 8.2)的 Na2HPO4。
丙烯酸、烯丙基胺、丙烯酰胺、N,N'-亚甲基双丙烯酰胺、3-甲基丙烯酰氧丙基三甲氧基硅烷(γ-MAPS,98%)购自山东鹏浩化工;N,N,N',N'-四甲基乙二胺(TEMED)、十二烷基硫酸钠(SDS)、过硫酸铵(APS)购自国药化学试剂公司(上海);其他试剂均为分析纯。
样品的配制:取鲜鸡蛋蛋清,溶于8倍体积的磷酸盐缓冲液,轻摇至蛋清溶解,过滤,4℃保存备用。
1.2 印迹、非印迹聚合物整体柱的制备
毛细管预处理方法:取一定长度的毛细管,分别用0.1 mol/L的NaOH溶液和蒸馏水依次冲洗40 min,注入γ-MAPS(50%体积分数的甲醇溶液)浸泡40 min,N2吹干,120℃恒温干燥3 h。

表1 单体溶液配比Table 1 Composition of the monomer solution (g/100 mL)
按照表1称取所需量的单体溶液溶于水,超声至完全溶解。用注射器将单体溶液分别引入毛细管中。封闭毛细管两端并置于-20℃冰箱中,24 h后取出置于50℃水浴中10 h。
1.3 整体柱的冲洗与印迹整体柱的再生
缓冲液使用前超声脱气10 min。整体柱每次实验前冲洗5~10 min。对比柱使用缓冲溶液冲洗,印迹柱使用含有3%SDS的缓冲溶液冲洗,并在90℃下至少保持15 min,之后再用缓冲溶液冲洗5 min,以彻底脱去印迹模板蛋白质。
2 结果与讨论
2.1 蛋白质分子待定模板印迹方法
理论上,在合成印迹聚合物时,将一个样品加入单体溶液中,若样品浓度高于某个值(印迹浓度阈值),则某些组分可作为模板分子被印迹。最先被印迹的是最高浓度组分,样品中某组分的浓度越高,被印迹的几率越高,反之亦然。样品浓度低于印迹浓度阈值则任何组分均不被印迹。通过调节样品的浓度,可以改变样品的印迹行为,即在样品浓度确定之前,被印迹的组分种类及数量均未确定。因此,该方法也称作“待定模板印迹”方法。
待定模板印迹方法对于复杂样品的分析具有特别意义。例如在蛋白质组学研究中,从体液、细胞或器官中提取的样品通常非常复杂,往往含有成百上千的蛋白质。从这些样品中除去高丰度组分,进而分析低浓度的生物标记物或特殊功能性分子,是一项耗费大量时间及经费的工作。在此情况下,待定模板印迹方法的出现将大大简化复杂样品的处理过程。
待定模板印迹的要点是组分部分印迹,即得到的聚合物对样品中高丰度组分具有特异亲和性。当从聚合物中除去模板分子,并再次作用于原样品时,印迹聚合物能选择性地吸附部分组分(模板),从而将样品分成两部分,一部分吸附于固体上,另一部分留在溶液中(图1)。通过调节样品的浓度,可以改变吸附、溶解两相组分的相对含量,从而得到一系列复杂性不同的“次级样品”,而每一“次级样品”中所含组分都比原样品简单。如果“次级样品”比较复杂,则可以将其作为新的“不定模板”,实施次级印迹(图1标出了第二代印迹)。理论上此过程可以一直进行,直到发现感兴趣的目标分子,或将其限制到一个足够简单的“馏分”中。
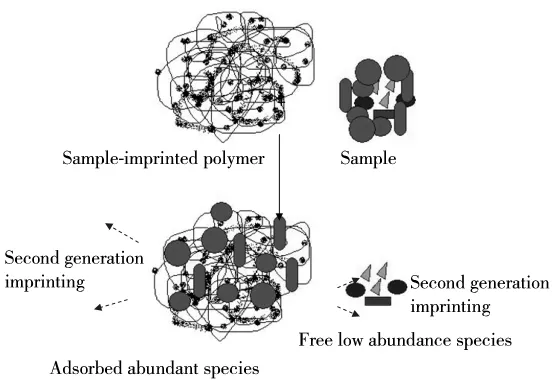
图1 蛋白质分子待定模板印迹聚合物与原样品的作用过程Fig.1 Schematic presentation of the interaction of the sample and imprinted polymer without standard templates
2.2 聚合物的特点
本文合成的聚合物是两性电解质冰胶,其特点是同时含有酸性和碱性基团,并具有超大孔结构。
同时使用酸、碱性单体制备蛋白质分子印迹聚合物的文献报道并不多见[23]。蛋白质分子由氨基酸聚合而成,本身是两性物质,因此作为印迹蛋白质分子的聚合物,两性电解质可比相似的酸性、碱性聚合物提供更多的相互作用位点,因而其印迹效果应该更加显著。实验发现聚合物与模板间的亲和性相当强,其洗脱液中必须添加SDS,且须在加热的条件下才能顺利实现脱附。另一方面,为了改善分离柱的通透性及提高分离速度,采用了具有超大孔结构的冰胶聚合物。

图2 聚合物整体柱的扫描电镜图Fig.2 SEM images of the polymers
单体溶液中,添加了比一般冰胶法含量多(~30%,表1)的交联剂N,N-亚甲基双丙烯酰胺。与丙烯酰胺相比,该交联剂疏水性稍强,形成的聚合物并非弹性胶状物,而是呈现蓬松多孔状结构。印迹与对照聚合物在外观上并无明显差异,只是印迹聚合物略显稠厚。图2是印迹聚合物(MIP)、对照聚合物(NIP)整体柱的扫描电镜图,外观上两种聚合物十分相似。
2.3 蛋白质在印迹、非印迹聚合物整体柱中的电泳
高丰度组分与单体相互作用,进而被印迹到聚合物中。当它们被除去后,聚合物中将留下亲和性空隙或位点,以此为基础的聚合物整体柱在样品分离中将显示出有趣的结果。
从图3可见,无论印迹柱还是对照柱(非印迹柱),都可实现鸡蛋清样品的快速分离。对照柱中电泳实验在16 min之内完成,图中清楚地呈现出6个以上的峰(图3A),代表样品中的高丰度组分。经与标准蛋白质电泳谱图对比,发现15 min附近的峰为卵清蛋白,2.5 min的峰为溶菌酶。由于标准品有限,其他谱峰未能定性。
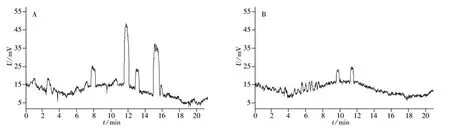
图3 鸡蛋清蛋白质在非印迹柱(A)与印迹柱(B)中的电泳图Fig.3 Electropherograms of chicken egg proteins on non-imprinted column(A)and sample-imprinted column(B)
对照样品在两根柱中的分离结果(图3),发现有几个主要蛋白质的峰信号出现消失或降低现象。除卵清蛋白(15 min)外,8、13 min处的谱峰消失,11.5 min处的峰高、峰面积大幅降低(图3B)。溶菌酶(2.5 min)的信号几乎消失。由此可见,印迹聚合物中存在针对高丰度组分的吸附位点,而这正是在聚合物的形成过程中高丰度组分的印迹行为造成的。虽然部分组分的谱峰降低或消失,但印迹柱中总的峰数目增加,主要集中在4~10 min之间(图3B)。这是由于印迹柱中存在特异性吸附位点,能够保留原来作为模板的部分高丰度组分,使其信号降低甚至消失;低丰度组分在单体溶液中浓度未达到印迹阈值,未发生印迹,聚合物中不存在相应特异性吸附位点,因此这些组分表现出快速迁移行为。在印迹聚合物柱中的进样量是对比柱中的2倍,所以部分低丰度组分的谱峰清晰可见(图3B,4.5~8 min)。
3 结论
待定模板印迹方法可以对复杂样品中的高丰度组分实施印迹而无需标准物质作为模板,形成的聚合物具有特异性吸附高丰度组分功能,从而为复杂样品的分离、分析提供了新的思路及操作技术。待定模板印迹聚合物通过捕获高丰度组分而增强低丰度组分的信号,这意味着它们可以用来从复杂样品中脱除高丰度组分,从而大幅度降低样品复杂性,便于进一步分析处理,且可方便地对低丰度组分进行浓缩富集。根据待定模板印迹原理可发展快捷、简便、低成本的样品处理及分离、分析方法,有望在复杂对象(如:蛋白质组学、医药、环境等)研究领域发挥重要作用。
[1]Wulff G,Gross T,Schonfeld R.Angew.Chem.Int.Ed.Engl.,1997,36(18):1962 -1964.
[2]Zhang W,He X W,Chen Y,Li W Y,Zhang Y K.Biosens.Bioelectron.,2011,26(5):2553-2558.
[3]Chen L X,Xu S F,Li J H.Chem.Soc.Rev.,2011,40(5):2922 -2942.
[4]Katz A,Davis M E.Nature,2000,403:286 -289.
[5]Chen Z Y,Xu L,Liang Y,Zhao M P.Adv.Mater.,2010,22(13):1488 -1492.
[6]Huang B Y,Chen Y C,Wang G R,Liu C Y.J.Chromatogr.A,2011,1218(6):849-855.
[7]Ou J,Li X,Feng S,Dong J,Dong X,Kong L,Ye M,Zou H.Anal.Chem.,2007,79(2):639 -646.
[8]Feng T,Hu Y F,Li G K.J.Instrum.Anal.(冯婷,胡玉斐,李攻科.分析测试学报),2011,30(10):1191-1198.
[9]Guo M J,Zhao Z,Fan Y G,Wang C H,Shi L Q,Xia J J,Long Y,Mi H F.Biomaterials,2006,27(24):4381 -4387.
[10]Liu J X,Yang K G,Deng Q L,Li Q R,Zhang L H,Liang Z,Zhang Y K.Chem.Commun.,2011,47(13):3969-3971.
[11]Li L,Chen Y G,Hu X X,Lu Y Y,Dai J J,Zhu Q H.J.Instrum.Anal.(李莉,陈粤冠,胡晓霞,卢瑶瑶,戴娇娇,朱全红.分析测试学报),2011,30(6):629-634.
[12]Hoshino Y,Koide H,Urakami T,Kanazawa H,Kodama T,Oku N,Shea K.J.Am.Chem.Soc.,2010,132(19):6644-6645.
[13]Saridakis E,Khurshid S,Govada L,Phan Q,Hawkins D,Crichlow G V,Lolis E,Reddy S M,Chayen N E.PNAS,2011,108(27):11081-11086.
[14]Kempe M,Mosbach K.J.Chromatogr.A,1995,691(1/2):317-323.
[15]Zhou Y M,Xu W G,Tong A J.Chin.J.Anal.Chem.(周艳梅,徐文国,童爱军.分析化学),2006,34(11):42-45.
[16]Zhou X,He X W,Chen L X,Li W Y,Zhang Y K.Chin.J.Anal.Chem.(周雪,何锡文,陈朗星,李文友,张玉奎.分析化学),2009,37(2):174-180.
[17]Yang H,Guo T Y,Zhou D Z.Int.J.Biol.Macromol.,2011,48(3):432-438.
[18]Shi H Q,Tsai W B,Garrison M D,Ferrari S,Ratner B D.Nature,1999,398:593-597.
[19]Nicholls I A,Rosengren J P.Bioseparation,2001,10(6):301-305.
[20]Yang H H,Zhang S Q,Tan F,Zhuang Z X,Wang X R.J.Am.Chem.Soc.,2005,127(5):1378-1379.
[21]Bereli N,Andac M,Baydemir G,Say R,Galaev I Y,Denizli A.J.Chromatogr.A,2008,1190(1):18-26.
[22]Bandow J E.Proteomics,2010,10(7):1416 -1425.
[23]Huang J T,Zhang J,Zhang J Q,Zheng S H.J.Appl.Polym.Sci.,2005,95(2):358-361.
