前列腺癌的分类及发生机制研究进展
2013-11-20朱圣生刘向云孙祖越复旦大学药学院上海200032
朱圣生 刘向云 孙祖越 (复旦大学药学院,上海 200032)
据统计,美国2010年有191 533例前列腺癌(PCa)新发病例和26 329例死亡病例,超过肺癌,居男性肿瘤之首〔1〕。占男性死亡原因的第二位。近十年来,PCa在我国的发病率呈明显上升趋势,已经成为发病率增长最快的肿瘤之一〔2〕。PCa分为激素依赖性PCa(HDPC)和激素非依赖性PCa(HIPC),原发性的PCa几乎都是典型的HDPC,雄激素是其关键性的生长因子。随着PCa病程的进展,所有的HDPC都会转变成HIPC,最终死于HIPC,这是 PCa发展的最终形式和必然结果〔3〕。本文对PCa的分类及HIPC的发病机制研究进行回顾性综述。
1 PCa的分类
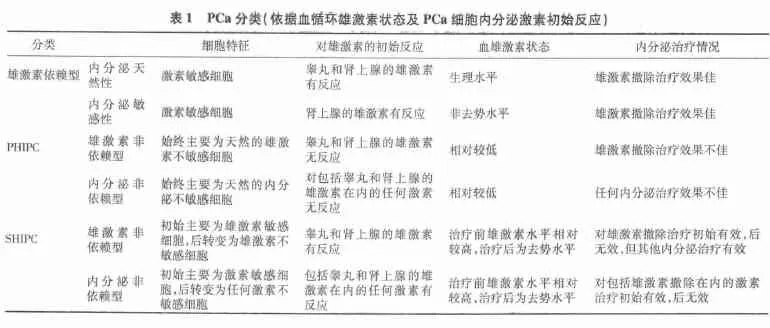
世界卫生组织分类方案根据PCa患者的血循环雄激素状态,将PCa分成雄激素依赖型和雄激素非依赖型〔3〕(见表1)。内分泌天然性PCa是指在睾丸和肾上腺源雄激素为生理水平而增殖形成的前列腺肿瘤。此类患者未接受任何激素治疗并且血雄激素为生理水平,在雄激素阻断下,这些肿瘤细胞会发生程序性凋亡;内分泌敏感性PCa是指接受一些不完全的雄激素撤除治疗,即接受不彻底的抗雄激素治疗如低剂量抗雄激素或雌激素单一治疗、放疗或前列腺切除前的新辅助治疗或间歇治疗,并且血清雄激素为非去势水平〔4〕。
根据Pca细胞初始时不同的激素依赖性,可将HIPC可以分成原发性激素非依赖型前列腺癌(PHIPC)和继发性激素非依赖型前列腺癌(SHIPC),以便更有利于HIPC的治疗选择和疗效判断。PHIPC是指PCa主要为天然的激素不敏感癌细胞,癌细胞的增殖和发展从一开始就不依赖任何内分泌激素,为雄激素非依赖性和内分泌不敏感性肿瘤,对任何内分泌治疗均无效。该类PCa又可进一步细分为原发性雄激素依赖型前列腺癌和原发性内分泌非依赖型PCa,为典型的内分泌难治性肿瘤;SHIPC是指PCa细胞由绝大多数天然的激素敏感细胞构成,癌细胞开始阶段依赖雄激素或其他内分泌激素生长和增殖,为雄激素依赖性或内分泌敏感性肿瘤。该类PCa初始对内分泌治疗有效,但一段时间后却发展为激素非依赖。该类PCa又可进一步细分继发性雄激素非依赖型PCa和继发性内分泌非依赖型 PCa〔4〕。
2 PCa的发生机制
2.1 雄激素受体(AR)相关机制
2.1.1 AR基因扩增 AR基因扩增是AR表达增加的有效机制。AR表达上调导致配基结合充足,可以在雄激素去势水平维持AR信号通路。Ruzi等〔5〕用荧光原位分析371例肿瘤标本,发现22%的转移性PCa和23%的局部复发性HIPC中有AR的扩增,而在原发灶中AR扩增率却低于2%。另外,Gregory等〔6〕研究发现,HDPC细胞转化为HIPC细胞以后,AR的表达增高,稳定性增加,且表达更加集中于细胞核内,肿瘤细胞对于双氢睾酮的促生长刺激作用更加敏感。此外,关于异种嫁接模型分析也表明由 HDPC到HIPC的过程中,AR基因表达增加,表明AR基因扩增在HIPC形成过程中起着重要的作用。
2.1.2 AR基因突变 AR基因突变是点突变,常见突变位点在C末端配基结合结构域(LBD)和铰链区〔7〕。AR的LBD突变使其对配基的选择性下降从而增加了能激活AR配体的种类。正常情况下,AR只能在与雄激素及双氢睾酮结合后才能活化,AR发生基因突变后,使得其他内源性激素或拮抗雄激素药物可发挥AR激动剂的作用,从而促进PCa细胞增殖。AR铰链区的突变使AR的转录活性增强,低浓度雄激素作用下就可以使靶基因激活。在PCa早期阶段,AR突变是比较少见的,但是随着恶性程度的增加,其发生率逐渐升高。Saraon等〔8〕对99例早期PCa患者进行分析,发现无一例AR编码序列有突变;而在进展期的38例PCa患者中,有8例AR发生突变,占21%。这些均表明AR的突变在PCa的进展中起重要作用。
2.1.3 AR的配体非依赖性激活 AR是一种磷蛋白,磷酸化状态的AR才具有转录活性,AR与某些因子结合而被激活或不依赖配体直接被激活称为AR的配体非依赖性激活。如生长因子〔表皮细胞生长因子(EGF)、纤维细胞生长因子(FGF)和内质细胞生长因子(KGF)等〕和细胞因子〔白介素-6(IL)-6、IL-4和IL-8等〕。生长因子为受体酪氨酸激酶的配基,受体型酪氨酸激酶的变化影响到AR通路的激活。研究证明,(HER-2/neu)基因是表皮生长因子受体(EGFR)的家族中的一员。LNCaP/HER-2细胞分泌的前列腺特异性抗原(PSA)比母系的人前列腺癌细胞(LNCaP)细胞分泌的PSA增高了6-7倍;在表达野生型AR的晚期PCa(LAPC4)-细胞中,HER2/neu基因在雄激素缺乏的情况下能增强 PSA增强子/启动子活性15倍〔9〕。另外,Her2/neu在PCa细胞中的表达较正常前列腺细胞中表达高,过表达的HER-2/neu不仅能促进LNCaP细胞的生长,而且对AR的转录激活作用不依赖雄激素的存在。由此推测,HER2/neu可以替代雄激素来促进PCa细胞生长。因此,HER2/neu途径可能是PCa进展的重要机制之一〔10〕。
血清中的胰岛素样生长因子(IGF)-1的水平可能与PCa的风险相关,通过降低IGF-1受体的表达,阻断其信号途径,在体内和体外均可抑制PCa细胞的生长。且在雄激素缺乏的情况下,IGF-1仍可促进AR的转录激活活性。IGF-1能活化野生型AR,这说明IGF-1引起AR活化可能是通过IGF-1配体受体结合后引起的信号级联途径实现的,而与AR是否突变无关。Himpe等〔11〕发现血清IGF-1增高的男性患PCa的风险是正常男性的4.3倍,然而,目前IGF-1的发挥此效应作用机制尚不明确。KGF亦有类似的作用。
细胞因子IL-6在PCa进展中发挥重要作用。临床表明,HIPC患者血清中IL-6水平较早期PCa患者血清IL-6水平相比显著升高,IL-6与其受体结合,同时激活(MAPK)和(STAT3)两条信号通道,进而激活AR信号途径〔12〕。同IL-6相似,HIPC患者的血清IL-4水平也明显增加。IL-4通过激活AR,上调雄激素阻断下的PCa细胞中PSA的表达,以及增强其与(ARE)的结合能力及AR核转运〔13〕。
?
2.1.4 AR共调节因子表达异常 AR共调节因子是能适当增强或降低靶基因转录活性、但不会改变其基础转录效率的一类蛋白质因子。根据其对AR转录效应的作用。可将其分为共激活、共抑制和双重作用因子。共调节因子本身不与DNA直接结合,不直接影响AR表达或改变AR进入细胞核的数量,而是通过其C端功能区与AR发生蛋白-蛋白间相互作用,改变AR在转录激活中对配体浓度的最低需求。使AR介导的转录活性大幅度改变。当AR与共激活因子作用时,可导致AR的异常活化从而大幅提高AR转录激活能力。反之,当AR与共抑制因子结合时,则导致AR转录活性的下降。二者平衡状态的丧失可以严重干扰靶基因的转录过程,最终导致HIPC发生〔14〕。目前,AR共调节因子对AR转录机制的影响尚不明确,但组蛋白及染色体结构的改变在转录调节过程中起主要作用。如甾体受体辅活化子1(SRC1)具有组蛋白乙酰基转移酶活性,而核受体辅抑制子(NCOR)等则具有去乙酰基转移酶活性,它们通过对染色体组蛋白的修饰,使染色体构象发生改变,影响其他共调节子的聚集,从而达到调节AR转录作用〔15〕。
SRC-1为甾体受体辅活子家族成员,其分子C-端的受体作用区有多个不同的LXXLL系列,介导SRC-1与不同核受体的配体结合区辅因子作用位点结合,通过自身的乙酰转移酶活性作用、并结合具有HAT活性的共激活子(CBP、P300等),使与DNA结合的组蛋白乙酰化,打断转录区域 DNA模板和组蛋白之间的连接,暴露目的基因,从而启动基因转录,并增强核受体介导的转录活性。体外实验表明,过表达SRC类共激活子可明显促进AR转录效应。这些共激活子还能促使抗雄激素类药物发挥雄激素样作用激活AR,或者协同其他类固醇激素活化AR,这是临床抗雄激素治疗的失败的重要原因。核受体辅阻遏子(NCoR)为核受体共抑制子超家族成员,其分子C-端受体作用区具有I/LXXII样的疏水系列,介导NCoR与核受体的LBD辅因子位点结合,通过发挥其自身的去乙酰基酶活性从而抑制核受体转录活性,而上述LBD辅因子位点又恰好与SRC-1等的AR作用位点相吻合,形成“共激活子/共抑制子”开关,二者的相对水平与核受体的活化状态密切相关。实验表明,在大部分HIPC细胞中SRC-1高表达,NCoR缺失表达;而在正常PCa细胞及HDPC细胞中NcoR过表达,这意味着由于共激活子和共抑制子的失衡,AR失去共抑制子的阻遏效应并在共激活子的刺激下,获得异常增强的AR转录活性,癌细胞在低浓度的雄激素环境下仍快速的增殖,从而引起HIPC的发生〔16〕。
2.2 PCa干细胞途径 PCa干细胞是可通过各种内分泌或旁分泌因子组成网络式调控信号通路实现其自我更新和具有高增殖潜力的中间细胞,PCa干细胞能够调控PCa从激素依赖转向激素非依赖,是HIPC发生的潜在的机制。现将其信号通路叙述如下。
2.2.1 SHh信号通路 前列腺组织的正常形态需要SHh信号通路,它可以调控干细胞的增殖。高度活化的SHh信号通路参与了PCa干细胞的起始,同时在PCa的转移中SHh也起到了重要的作用。在PCa干细胞中,SHh高度活化。实验表明在前列腺干细胞中SHh的活化导致了PCa干细胞的产生〔17〕。
2.2.2 Wnt/β-Catenin信号通路 Wnt/β-Catenin 信号促进癌细胞增殖和分化,Wnt通过阻止β-Catenin的分解而激活Tef/lef,Tef/lef调节转录因子来促进细胞的分化。抑制Wnt信号通路,癌干细胞样细胞的自我更新能力明显降低,这表明Wnt信号通路参与了PCa干细胞的产生〔18〕。
2.2.3 Notch信号通路 研究表明Notch受体和配体不仅存在于正常的前列腺干细胞也存在于PCa细胞。高活性的及失调的Notch信号通路将会使前列腺干细胞转化为PCa干细胞。在PCa干细胞中 Notch-1呈过高表达,目前把Notch-1作为PCa干细胞的表面标志物之一〔19〕。
2.2.4 转换生长因子(TGF)-β信号通路 正常前列腺组织中静止的干细胞是受严格调控的。前列腺导管附近的组织中含有正常的干细胞,这些干细胞表达TGF-β,同时TGF-β能够抑制导管附近细胞的增殖。如果TGF-β与其他的自分泌因子的平衡被打破,这些细胞就表现出较强的增殖能力,转化为PCa干细胞〔20〕。
2.2.5 PTEN/PI3K/Akt信号通路 Dubrovska等〔21〕发现 Sca-1/Bcl-2/CK5/p63+前列腺干细胞和它的分化亚型Sca-1/Bcl-2/CK5/p63-细胞中缺乏磷酸脂酶,这说明PTEN/PI3K/Akt信号通路参与了前列腺干细胞的调控。Dubrovsk等〔22〕在研究CD133+/CD44+PCa干细胞样细胞的功能时也发现PTEN/PI3K/Akt信号通路在维持PCa干细胞样细胞的自我更新和分化方面都具有重要的作用。
2.3 其他机制
2.3.1 前列腺组织内雄激素的合成 在拮抗雄激素治疗或者去除雄激素后,PCa细胞可通过调节前列腺内的雄激素合成来对抗机体血液循环中雄激素下降而存活。Montgomery等〔23〕也发现HIPC肿瘤组织中的雄激素浓度是未进行雄激素治疗前的PCa组织中的3.8倍。Glinskii等〔24〕对发生骨转移晚期 PCa和早期的PCa进行对比,发现在骨转移晚期PCa标本中参与雄激素合成代谢类基因表达水平明显增高。比如 3βHSD2,AKR1C3,SRD5A1,AKR1C1,UGT2B15 等。这些基因的蛋白产物均参与雄激素合成的各个过程。实验表明〔25〕,PCa组织中的胆固醇含量较正常组织明显增高,并且大部分处于游离状态,而前列腺内的雄激素可由胆固醇或其他广泛存在的前体分子重新合成,比如脱氢表雄酮(DHEA)。许多编码生成类固醇的酶基因,包括 3βHSD2,AKR1C3,SRD5A1,CYP17A1,CYP19A 1和U BT 2B15都能调节DHEA的合成。尤其是AKR1C3可在前列腺中将雄烯二酮转化为睾酮。而3βHSD2可将 DHEA转化为一种睾酮的底物雄烯二酮。而SRD5A1可将睾酮转化为高亲和力的双氢睾酮(DHT)。与未接受治疗的PCa患者相比,去势后雄激素非依赖型PCa中参与雄激素代谢调节的基因表达明显上调。这表明前列腺内的雄激素合成在PCa的进展中起重要作用。
2.3.2 AR外的旁路途径 细胞凋亡是指细胞在一定生理或病理条件下,遵循自身的程序主动死亡过程。细胞凋亡受多种基因调控,正常情况下PCa细胞凋亡而使临床上患者的病情缓解,但一旦细胞凋亡的调控基因发生表达异常,肿瘤细胞不但不会凋亡反而继续生长、增殖致使病情恶化。参与细胞凋亡调控的主要包括抑制因子(bcl-2,人核转录因子(NF)-kB,IAPs等)和诱导因子(p53,FAS/FASL,caspases家族、Bax等)。抑制因子的表达增强与诱导因子的基表达下降与肿瘤的发生发展有关。在PCa的有关细胞凋亡研究中,bcl-2是主要的抗细胞凋亡因子。bcl-2蛋白在正常的前列腺基底细胞表达,但在分泌性上皮细胞中不表达;而在多数中、低级PCa中也发现bcl-2不表达,但在HIPC中表达明显。Bcl-2表达水平越高,肿瘤恶性程度越高〔26,27〕。
野生型p53是一种抑癌基因,野生型P53蛋白在维持细胞正常生长、细胞周期调节和抑制恶性增殖中发挥重要作用。野生型p53还是一种生长抑制性蛋白,可以抑制正常和转化细胞的生长,使细胞停滞在G1期。当细胞DNA损伤时,p53对其细胞周期进行负调控,抑制G1晚期的基因转录,以保证基因的稳定表达和细胞分裂的正常进行。CWR22大鼠模型实验表明,去势后p53表达显著降低,从而丧失其对细胞周期的控制作用,这促进了去势治疗后的PCa生长、恶化。另外,抑癌基因p53在HIPC中突变率很高。当p53突变后,由于空间构象改变,影响到转录活化功能及P53蛋白磷酸化过程,这不但失去了野生型p53抑制肿瘤增殖作用,而且突变本身又使该基因具备癌基因功能。突变P53蛋白与野生型P53蛋白结合,形成的这种寡聚蛋白不能结合DNA,使得一些癌基因转录失控,促进PCa的生长、恶化〔28~30〕。
前列腺主要由分泌性腺上皮细胞、基底细胞和NE细胞组成。基底细胞在雄激素作用下可转化为分泌性腺上皮细胞,二者均表达AR,而NE细胞一般不表达AR,NE细胞可分泌多种活性物质,包括PTHRP、神经递质5-羟色胺、神经肽类激素等。这些物质通过间质上皮相互作用机制来调节前列腺上皮细胞的生长〔31〕。在PCa的发生发展中,NE细胞起着至关重要的作用。研究发现,HIPC中NE细胞显著增多,患者血清中NE细胞分泌产物的升高程度与患者预后密切相关。体外研究发现,PCa细胞株在无甾体激素的条件下培养,PCa细胞可转化为NE细胞样细胞。这些细胞可分泌NE细胞的一些产物,其增殖不依赖雄激素。因此,推测在去势治疗中,神经内分泌细胞并不受去势治疗的影响,在低雄激素的环境下,NE细胞通过其神经内分泌产物而使周围的癌细胞增生,使肿瘤逐渐逃避激素治疗〔32〕。神经内分泌细胞样分化作用及细胞凋亡调控异常在HIPC的发生发展中发挥了重要作用。
3 展望
HIPC的发生是一个涉及多种机制的复杂过程,临床上不同个体的发生机制也不尽相同。HIPC发病机制的阐明能为临床治疗提供理论依据,并能根据不同信号途径进行靶向药物设计。今后以下几个方向进行研究。首先,发现能预测PCa预后和有HIPC倾向的最敏感的生物标志物,并针对其设计出快速,灵敏,可靠的检测方法;其次,加大对HIPC的发病机制的研究力度,找出特异性高的治疗靶点;最后,根据HIPC的不同发生机制,设计分子靶向治疗药物。
1 Siegel R,Naishadham D,Jemal A.Cancer statistics,2012〔J〕.CA Cancer J Clin,2012;62(1):10-29.
2 孙颖浩.前列腺癌诊治进展〔J〕.上海医学,2011;34(7):487-8.
3 Newling D,Fossa SD,Andersson L,et al.Assessment of hormone refractory prostate cancer〔J〕.Urology,1997;49(4A Suppl):46-53.
4 杨国胜,陈昭典.激素抵抗型前列腺癌分类及机制的进展〔J〕.中国男科学杂志,2004;18(2):6-10.
5 Ruiz C,Holz DR,Oeggerli M,et al.Amplification and overexpression of vinculin are associated with increased tumour cell proliferation and progression in advanced prostate cancer〔J〕.J Pathol,2011;223(4):543-52.
6 Titus MA,Tan JA,Gregory CW,et al.14-3-3{eta}amplifies androgen receptor actions in prostate cancer〔J〕.Clin Cancer Res,2009;15(24):7571-81.
7 Mellado B,Codony J,Ribal MJ,et al.Molecular biology of androgen-independent prostate cancer:the role of the androgen receptor pathway〔J〕.Clin Transl Oncol,2009;11(1):5-10.
8 Saraon P,Jarvi K,Diamandis EP.Molecular alterations during progression of prostate cancer to androgen independence〔J〕.Clin Chem,2011;57(10):1366-75.
9 Dahl M,Bouchelouche P,Kramer-Marek G,et al.Sarcosine induces increase in HER2/neu expression in androgen-dependent prostate cancer cells〔J〕.Mol Biol Rep,2011;38(7):4237-43.
10 Hsu FN,Yang MS,Lin E,et al.The significance of Her2 on androgen receptor protein stability in the transition of androgen requirement in prostate cancer cells〔J〕.Am J Physiol Endocrinol Metab,2011;300(5):E902-08.
11 Himpe E,Potikanond S,Verdood P,et al.Attenuation of IGF-I receptor signaling inhibits serum-induced proliferation of prostate cancer cells〔J〕.Growth Horm IGF Res,2011;21(5):252-9.
12 Wegiel B,Evans S,Hellsten R,et al.Molecular pathways in the progression of hormone-independent and metastatic prostate cancer〔J〕.Curr Cancer Drug Targets,2010;10(4):392-401.
13 Roca H,Craig MJ,Ying C,et al.IL-4 induces proliferation in prostate cancer PC3 cells under nutrient-depletion stress through the activation of the JNK-pathway and survivin upregulation〔J〕.J Cell Biochem,2011;113:1569-80.
14 Mehraein-Ghomi F,Basu HS,Church DR,et al.Androgen receptor requires JunD as a coactivator to switch on an oxidative stress generation pathway in prostate cancer cells〔J〕.Cancer Res,2010;70(11):4560-8.
15 Shiota M,Yokomizo A,Tada Y,et al.Peroxisome proliferator-activated receptor gamma coactivator-1alpha interacts with the androgen receptor(AR)and promotes prostate cancer cell growth by activating the AR〔J〕.Mol Endocrinol,2010;24(1):114-27.
16 Agoulnik IU,Weigel NL.Coactivator selective regulation of androgen receptor activity〔J〕.Steroids,2009;74(8):669-74.
17 Kim TJ,Lee JY,Hwang TK,et al.Hedgehog signaling protein expression and its association with prognostic parameters in prostate cancer:a retrospective study from the view point of new 2010 anatomic stage/prognostic groups〔J〕.J Surg Oncol,2011;104(5):472-9.
18 Wan X,Liu J,Lu JF,et al.Activation of beta-catenin signaling in androgen receptor-negative prostate cancer cells〔J〕.Clin Cancer Res,2012;18(3):726-36.
19 Wang Z,Li Y,Banerjee S,et al.Down-regulation of Notch-1 and Jagged-1 inhibits prostate cancer cell growth,migration and invasion,and induces apoptosis via inactivation of Akt,mTOR,and NF-kappaB signaling pathways〔J〕.J Cell Biochem,2010;109(4):726-36.
20 Fuzio P,Ditonno P,Rutigliano M,et al.Regulation of TGF-beta1 expression by androgen deprivation therapy of prostate cancer〔J〕.Cancer Lett,2012;318(2):135-44.
21 Dubrovska A,Elliott J,Salamone RJ,et al.Combination therapy targeting both tumor-initiating and differentiated cell populations in prostate carcinoma〔J〕.Clin Cancer Res,2010;16(23):5692-702.
22 Dubrovska A,Kim S,Salamone RJ,et al.The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations〔J〕.Proc Natl Acad Sci U S A,2009;106(1):268-73.
23 Montgomery RB,Mostaghel EA,Vessella R,et al.Maintenance of intratumoral androgens in metastatic prostate cancer:a mechanism for castration-resistant tumor growth〔J〕.Cancer Res,2008;68(11):4447-54.
24 Glinskii OV,Sud S,Mossine VV,et al.Inhibition of prostate cancer bone metastasis by synthetic TF antigen mimic/galectin-3 inhibitor lactulose-L-leucine〔J〕.Neoplasia,2012;14(1):65-73.
25 Moltzahn F,Thalmann GN.Bone metastasis in prostate cancer〔J〕.Urologe A,2012;51(1):20-6.
26 Anvari K,Seilanian TM,Kalantari M,et al.Expression of Bcl-2 and Bax in advanced or metastatic prostate carcinoma〔J〕.Urol J,2012;9(1):381-8.
27 钟恩宏,孙祖越,顾 正,等.依立雄胺对前列腺癌细胞(LNCaP)前列腺特异性抗原和Bcl-2蛋白体外表达的抑制作用〔J〕.中国药理学与毒理学杂志,2003;17(1):29-34.
28 Liu R,Zhang Z,Xu Y.Downregulation of nucleostemin causes G1 cell cycle arrest via a p53-independent pathway in prostate cancer PC-3 cells〔J〕.Urol Int,2010;85(2):221-7.
29 Chen H,Sun Y,Wu C,et al.Pathogenesis of prostatic small cell carcinoma involves the inactivation of the P53 pathway〔J〕.Endocr Relat Cancer,2012;19(3):321-31.
30 刘向云,杨荣富,胡文娟,等.人OC-3-VGH卵巢癌细胞裸小鼠肿瘤模型的建立〔J〕.中国实验动物学报,2009;17(2):108-10.
31 姜永光,罗 勇.前列腺癌雄激素依赖特性转变的生物学机制的研究进展〔J〕.现代泌尿外科杂志,2008;13(6):411-4.
32 Danza G,Di Serio C,Rosati F,et al.Notch signaling modulates hypoxiainduced neuroendocrine differentiation of human prostate cancer cells〔J〕.Mol Cancer Res,2012;10(2):230-8.
