“硅替代”新药创制研究进展
2013-10-28叶婷婷王秋岩沈凌宏吴慧丽殷晓浦
叶婷婷,王秋岩,沈凌宏,曹 丹,吴慧丽,殷晓浦
(1. 杭州师范大学生命与环境科学学院,浙江 杭州 310036;2. 杭州师范大学生物医药与健康研究中心,浙江 杭州 311121)
“硅替代”新药创制研究进展
叶婷婷1,王秋岩2,沈凌宏1,曹 丹2,吴慧丽2,殷晓浦2
(1. 杭州师范大学生命与环境科学学院,浙江 杭州 310036;2. 杭州师范大学生物医药与健康研究中心,浙江 杭州 311121)
“硅替代”新药创制即合成现有药物的含硅结构类似物.含硅药物与其碳结构类似物相比,具有独特的理化性质,并表现出特定的生物活性,如更强的药效、更高的选择性和更小的毒副作用.因此,对现有药物进行硅替代是设计、改造药物的有效途径.文章对各种硅替代药物进行了全面综述.
硅替代;含硅药物;药物设计
“硅替代”新药创制是指合成现有药物的含硅结构类似物.硅和碳有许多相似之处,它们是同属第IV主族的元素,与其它元素原子都形成4个共价键.然而,这两个元素也存在一些差异,由于原子大小、电负性、亲脂性以及化学特性的不同(表1),使得硅替代新药具有许多优异的性质,如更强的药效、更高的选择性和更小的毒副作用[1-2].设计新型含硅药物可以产生有效的生物活性,并作为新药申请专利保护,为新药研究提供了一条新途径[3-5].

表1 C和Si原子性质比较Tab. 1 Properties of carbon and silicon atoms
1 硅替代后药效提高的药物
含硅药物比其碳结构类似物的亲脂性更高,从而影响药物体内代谢.亲脂性高的硅替代药物能显著增加药物分布容积,具有良好的组织渗透能力;还能增加表观油水分配系数,增强药物血脑屏障的通透性,进而增强中枢神经系统活动.因此,硅替代药物可以延长药效作用的持续时间进而提高药效.
1.1 硅-氯贝丁酯(Sila-Clofibrate)
氯贝丁酯(Clofibrate)为降血脂药,是过氧化物酶体增生因子激活受体α(peroxisome pro-liferators-activated receptors α)的激动剂,同时还具有抗炎作用[6-7].
用三甲基硅烷基团替代氯贝丁酯苯氧基上氯原子,合成得到硅-氯贝丁酯(结构式1).角叉菜胶(Carrageenan)具有明显的诱发炎症作用,可用于抗炎药物的筛选和研究.Ziaee等[8]通过角叉菜胶诱发炎症的空气囊模型来分析氯贝丁酯和硅-氯贝丁酯的抗炎活性:在小鼠背部皮下注射无菌空气形成空气囊.将小鼠分为7组,每组7只,各给药组通过强饲口服分别给予等量的氯贝丁酯和硅-氯贝丁酯(2.5,5,10 mg/kg),对照组给予等量生理盐水.给药后1 h,在各组小鼠空气囊中注射4 mL溶解在生理盐水中的1%角叉菜胶.然后提取囊液,分析囊液中总白细胞量.表2显示硅-氯贝丁酯给药组的小鼠囊液中白细胞量明显少于氯贝丁酯给药组,这表明:硅-氯贝丁酯增加了亲脂性,改善了药效作用,抗炎效果得到了加强.

结构式1 硅-氯贝丁酯Structure 1 Sila-Clofibrate

表2 氯贝丁酯和硅-氯贝丁酯对小鼠囊液中白细胞量的影响
1.2 Karenitecin(BNP 1350)

结构式2 Karenitecin (BNP 1350)Structure 2 Karenitecin (BNP 1350)
喜树碱(Camptothecin)是一种细胞毒性喹啉类生物碱,能抑制DNA拓扑异构酶,具有显著抗肿瘤活性,为一种临床上常用的抗癌药物.然而,喜树碱在临床试验中有高度的药物不良反应特性[9-10].
喜树碱类药物已在临床得到了广泛应用,但是由于生物利用度低,稳定性差,不能用于神经胶质瘤的治疗.增加药物的亲脂性,将有助于这一问题的解决.基于这一设想,BioNumerik公司的研究人员合成了用
硅烷基取代喜树碱7位碳原子的化合物Karenitecin(BNP 1350,结构式2)[9].
Karenitecin 血浆稳定性、抗癌活性较喜树碱都有较大的改善,而且具有高度亲脂性,使得药物容易跨过血脑屏障.研究表明 Karenitecin 在治疗神经胶质瘤方面具有广阔的前景.体外抑酶活性实验结果表明,该产品具有广谱抗肿瘤活性.利用该产品治疗转移性黑素瘤的Ⅱ期临床试验结果显示,大部分病人病情得到控制,而且耐受性良好[11].在对复发或难治性非小细胞肺癌患者进行的Ⅱ期临床试验结果显示,43%的复发患者和50%的难治性患者的病情得到控制[12].该产品与抗肿瘤药马磷酰胺联用时,对白血病Molt-4、成神经管细胞瘤D283、成神经管肿瘤SK-N-SH等细胞株有协同增效的抗癌作用[13].目前,该产品与已上市的喜树碱类药物拓扑替康(Topotecan)联合治疗晚期上皮卵巢癌的Ⅲ期临床试验以及口服给药治疗实体瘤和肺癌的Ⅰ期临床试验已经在进行.IC50是指细胞被抑制一半时抑制剂的浓度,可以用来衡量药物抑制细胞的能力,该数值越低,药物抑制作用越强.研究人员对喜树碱类药物作用于人体卵巢癌细胞A2780的抑制作用(表3)进行的研究表明,BNP 1350在治疗人体卵巢癌方面效果较好[14-15].毒理学研究表明,Karenitecin具有可逆的中性白细胞减少和血小板减少的毒性,但没有可累积的骨髓抑制毒性,也没有喜树

表3 喜树碱类药物对卵巢癌细胞A2780的抑制作用Tab. 3 The inhibition of camptothecins forovarian cancer cells
碱类化合物常见的严重的腹泻反应[14].
2 硅替代后选择性提高的药物
在药物中引入硅基团后,可影响含有某些官能团(如—OH等)的药物对受体的亲和力.硅比碳的电负性小,因此硅醇的氢键键能更大.药物的甲醇官能团作为氢键供体,与该药对受体的亲和力有关.若用硅醇官能团替代药物的甲醇官能团,可增强药物对受体的亲和力,提高药物作用的选择性,即作用专一性强[16].同时,硅替代后的某些官能团所能发生的化学反应也不相同,可能影响药物在体内的性质.
2.1 硅-文法拉辛(Sila-Venlafaxine)

结构式3 硅-文法拉辛Structure 3 Sila-venlafaxine
文法拉辛(Venlafaxine)是一种抗抑郁药,它是第一个去甲肾上腺素(noradrenaline)和5-羟色胺(serotonin)再摄取的双重抑制剂.文法拉辛主要药理机制为抑制神经突触前膜对5-羟色胺和去甲肾上腺素的再摄取,增强中枢神经系统5-羟色胺和去甲肾上腺素神经递质的活性,发挥抗抑郁作用,也可阻断多巴胺(dopamine)的再摄取[17-18].
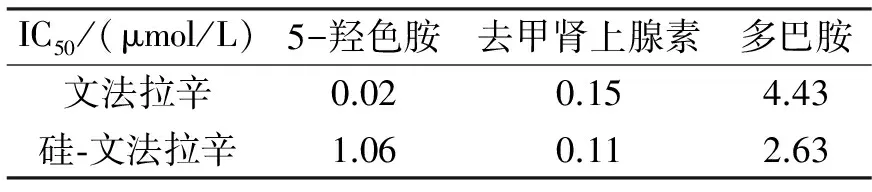
表4 文法拉辛和硅-文法拉辛对5-羟色胺、去甲肾上腺素和多巴胺再摄取的抑制作用Tab. 4 The inhibition of venlafaxine and sila-venlafaxinefor serotonin, noradrenaline and dopamine
通过合成得到硅-文法拉辛(结构式3), 其结构中原文法拉辛的甲醇官能团替代为硅醇官能团,二者理化性质非常相似.通过IC50值的分析来对比硅-文法拉辛和文法拉辛阻断去甲肾上腺素、5-羟色胺和多巴胺再摄取的抑制作用(表4).结果表明,硅-文法拉辛对去甲肾上腺素和多巴胺再摄取的抑制作用有所增强,对5-羟色胺再摄取抑制作用明显降低[19].硅-文法拉辛对去甲肾上腺素再摄取的抑制作用比对其他两种神经递质的抑制作用强约10倍.因此,硅-文法拉辛是选择性的去甲肾上腺素再摄取抑制剂,比作为去甲肾上腺素和5-羟色胺再摄取的双重抑制剂的文法拉辛,提高了药物作用的选择性.硅-文法拉辛由于硅原子的引入,增强了药物对去甲肾上腺素的亲和力,提高了作用的选择性,增强作用的专一性,药理作用发生了改变[20-21].
2.2 氟-硅-六氢地芬尼多(p-fluoro-hexahydro-sila-difenidol,pFHHSiD)
盐酸地芬尼多(Difenidol)对痉挛的血管有扩张作用,能调整前庭神经的异常冲动,抑制呕吐中枢及改善眼球震颤,可用于眩晕、呕吐的患者.地芬尼多只有轻度抗毒蕈碱型胆碱受体(muscarinic receptor,M受体)作用[22].
氟-硅-六氢地芬尼多(p-fluoro-hexahydro-sila-difenidol,pFHHSiD,结构式 4)是硅替代药物中很成功的例子,它是新型M受体阻断药,具有很好的利用价值[23-24].阻断药的pA2值是指能使激动药在提高一倍浓度后,仍产生原浓度作用强度时所需阻断药摩尔浓度的负对数值,常用以表示受体阻断药的作用强度,pA2值越大,药物的阻断作用越大.通过pA2值分析胆碱能M受体阻断药阿托品(Atropine)、哌仑西平(Pirenzepine)、pFHHSiD拮抗乳内动脉乙酰胆碱M受体的活性(表5),结果表明,pFHHSiD对M受体有高度亲和力,具有较强的拮抗M受体的活性[23].

结构式4 氟-硅-地芬尼多Structure 4 p-Fluoro-sila-difenidol

表5 M受体阻断药阿托品、哌仑西平、氟-硅-六氢地芬尼多对乳内动脉乙酰胆碱M受体的拮抗活性
3 硅替代后降低毒性的药物
大多数的毒理学试验数据证明含硅化合物的毒性较低,硅替代后的新药提高了药物的安全性[25].
氟哌啶醇(haloperidol)是一种抗精神病药,通过阻断多巴胺D2受体发挥作用.多巴胺功能亢进与妄想、幻觉和思维障碍等精神病症状相关.
Tacke等[26-28]用硅替代了氟哌啶醇哌啶环上的碳,合成得到硅-氟哌啶醇.氟哌啶醇和其类似物硅-氟哌啶醇对重组人多巴胺D2受体的亲和力分析表明,硅-氟哌啶醇比氟哌啶醇的亲和力强约5倍,这是由于硅醇的氢键键能增大导致的,其对药理作用的影响目前还不清楚.
除了亲和力增强,体内代谢研究表明,硅-氟哌啶醇与氟哌啶醇的代谢途径不同,从而避免了氟哌啶醇有毒副作用的代谢物产生,因此硅替代后该药明显降低了毒副作用[29-30].研究人员通过电喷雾质谱(ESI-MS)分析研究出药物代谢过程(图1,图2).氟哌啶醇的吡啶代谢产物有严重的神经毒性,可能产生类似于帕金森症的副作用.然而由于Si—O化学键的键能比C—O键的键能大以及Si=C键在生理条件下很不稳定,硅基吡啶代谢物不会产生.硅-氟哌啶醇在C—N位经过氧化还原反应形成代谢中间体,进而开环最终形成了代谢产物硅二醇[30].硅替代药物不同的代谢路径减少药物毒副产物的生成,从而降低药物毒性,为新药的研究提供了一种思路.

图1 氟哌啶醇的代谢过程Fig. 1 Metabolic process of haloperidol

图2 硅-氟哌啶醇的代谢过程Fig. 2 Metabolic process of sila-haloperidol
4 通过硅替代设计的新药
硅比碳稳定,硅替代新药中引入的硅基对化合物的活性基团起保护作用,有提高药物的稳定性和延长药效等优势,因此硅替代为新药开发提供了新思路,并且应用于药物设计[31].
4.1 氟硅唑(Flusilazole)和氟硅菊酯(Silafluofen)
氟硅唑(Flusilazole,结构式5)是一种市场上广泛使用的农用杀菌剂,性质稳定,对哺乳动物和鱼等水生生物毒性低.氟硅唑潜在的功效已被应用于人体,临床和毒理学试验证明了该化合物自身无毒性[32].
氟硅菊酯(Silafluofen,结构式6)是广泛应用的农用杀虫剂,其杀虫谱广,对哺乳动物和鱼类低毒,还具有高效、低残留、性质稳定等优点.氟硅菊酯急性经口、皮LD50(大鼠)>5 000 mg/kg;吸入LC50(大鼠/4 h)>6 610 mg/L;其性质稳定,常温储藏两年,在碱性条件下不易分解[33].缪康等[34]研究了氟硅菊酯等农药对秧田灰飞虱的防治效果.灰飞虱是主要的水稻害虫之一,由于长期、单一、大规模地使用防治农药如毒死蜱,灰飞虱已经产生了抗药性.拟除虫菊酯类醚菊酯和氟硅菊酯都是神经毒剂,毒副作用极低.田间杀虫实验表明:二者的防治效果显著好于毒死蜱,具有速效性好、持效期长的优点;在相同用量下,氟硅菊酯较醚菊酯的防治效果更好、持效期更长.

结构式5 氟硅唑Structure 5 Flusilazole
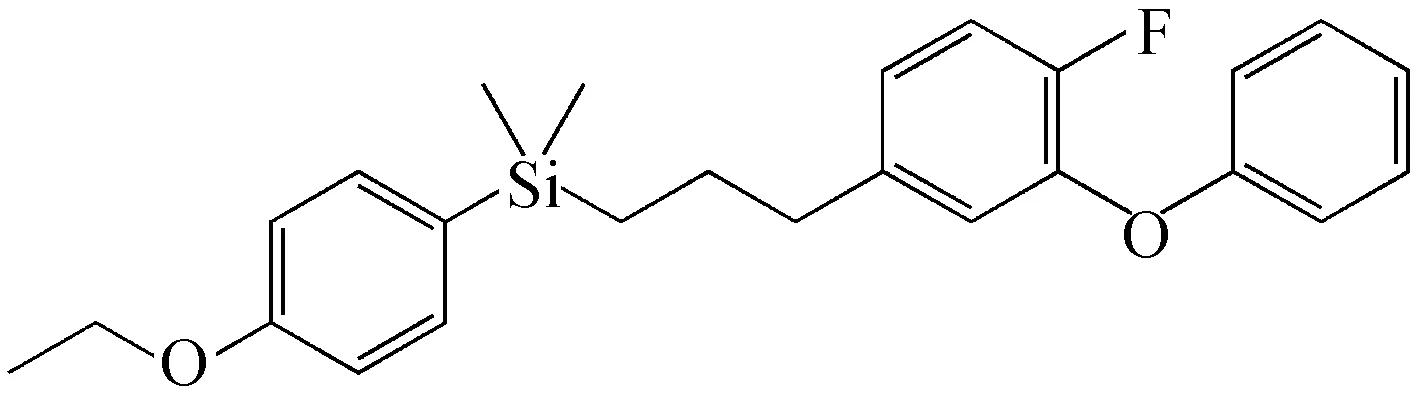
结构式6 氟硅菊酯Structure 6 Silafluofen
在目前高毒农药危害严重、常规药剂已经产生严重抗性等诸多情况下,氟硅唑和氟硅菊酯等低毒硅替代农药的应用可大大减少高毒农药在农业上的使用,减轻对人体健康和生态环境的影响,近年来已取得了较大的成功.
4.2 硅醇类
酰胺类和酯类在水解酶如蛋白酶的作用下形成具有两个羟基的四面体中间体,进而分别水解成羧酸和胺类物质,以及羧酸和醇类物质.抑制这一类水解酶的作用,从而起到治疗作用是药物设计的有效途径.硅原子替代四面体中间体中不稳定的碳原子后,该四面体中间体就可以成为稳定的化合物而不被进一步水解.硅二醇(Silandiol)和硅三醇(Silantriol)就是此类稳定的四面体中间体,是酰胺类和酯类化合物水解中间体的类似物.硅二醇和硅三醇与蛋白酶结合后可以抑制水解作用,作为蛋白酶抑制剂[35-36]类的药物发挥疗效.
目前,已得到几种酶的硅二醇抑制剂,有较强的疗效.含硅二醇结构的化合物(结构式7)可以很好地抑制人类免疫缺陷病毒(HIV)蛋白酶的活性,其酶亲和活性浓度Ki值为2.7 nmol/L,半数抑制浓度IC50为14 nmol/L,将会成为用于治疗艾滋病(AIDS)的一种新药[37].硅三醇化合物(结构式8)也可以作为血管紧张素转化酶抑制剂,并且其毒性很低,对ACE酶的IC50值为121 μmol/L[38].

结构式7 硅二醇结构的化合物Structure 7 Silandiol compound
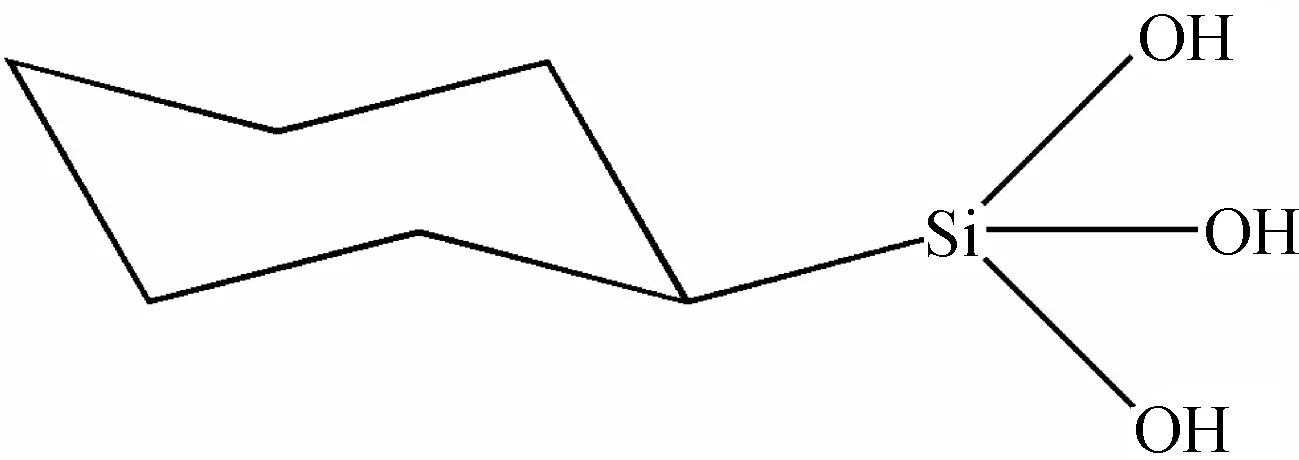
结构式8 硅三醇结构的化合物Structure 8 Silantriol compound
5 展 望
增强药物疗效、提高药物作用专一性、提高药物安全性是药物研究中亟待解决的重要课题.硅替代后的药物由于其优良的特性,为药物设计提供了很好的思路,研究人员已经通过对药物芳基核心结构修饰以及用硅烷基或含硅基团如硅醇替代等途径,设计了药效强、专一性高且毒副作用小的新型含硅药物,同时进行了含硅药物在药理及毒理学等方面的研究.虽然硅替代新药创制的研究还处于起步阶段,还有很多问题如合成含硅药物的技术、反应路线、制备成本、成键稳定性等问题有待解决,但是前景十分广阔,硅替代新药创制已经逐渐成为现代医药领域中很有前途、极为活跃的研究领域.
[1] Bains W, Tacke R. Silicon chemistry as a novel source of chemical diversity in drug design[J]. Current Opinion in Drug Discovery & Development,2003,6(4):526-543.
[2] Mills J S, Showell G A. Exploitation of silicon medicinal chemistry in drug discovery[J]. Expert Opinion on Investigational Drugs,2004,13(9):1149-1157.
[3] Shimizu M, Hiyama T. Silicon-bridged biaryls: molecular design, new synthesis, and luminescence control[J]. Synlett,2012,23(7):973-989.
[4] Showell G A, Mills J S. Chemistry challenges in lead optimization: silicon isosteres in drug discovery[J]. Drug Discovery Today,2003,8(12):551-556.
[5] Tacke R, Metz S. Odorant design based on the carbon/silicon switch strategy[J]. Chemistry & Biodiversity,2008,5(6):920-941.
[6] Brown J D, Plutzky J. Peroxisome proliferator-activated receptors as transcriptional nodal points and therapeutic targets[J]. Circulation,2007,115(4):518-533.
[7] Tziomalos K, Athyros V G, Karagiannis A,etal. Anti-inflammatory effects of fibrates: an overview[J]. Current Medicinal Chemistry,2009,16(6):676-684.
[8] Ziaee M, Samini M, Bolourtchian M,etal. Synthesis of a novel siliconized analog of clofibrate (silafibrate) and comparison of their anti-inflammatory activities[J]. Iran J Pharm Res,2011,11(1):91-95.
[9] 宋云龙,张万年,季海涛,等.DNA拓扑异构酶I结构、功能及喜树碱类抗癌药物研究进展[J].中国药学杂志,2002,37(9):646-650.
[10] 黄敏,丁健.拓扑异构酶Ⅰ抑制剂研究进展[J].中国新药杂志,2007,16(13):990-1000.
[11] Munster P N, Daud A I. Preclinical and clinical activity of the topoisomerase I inhibitor, karenitecin, in melanoma[J]. Expert Opinion on Investigational Drugs,2011,20(11):1565-1574.
[12] Miller A A, Herndon J E, Gu L,etal. Phase II trial of karenitecin in patients with relapsed or refractory non-small cell lung cancer(CALGB 30004)[J]. Lung Cancer,2005,48(3):399-407.
[13] Grossman S A, Carson K A, Phuphanich S,etal. Phase I and pharmacokinetic study of karenitecin in patients with recurrent malignant gliomas[J]. Neuro-Oncology,2008,10(4):608-616.
[14] Hattum A H, Schlüper H M M, Hausheer F H,etal. Novel camptothecin derivative BNP1350 in experimental human ovarian cancer: determination of efficacy and possible mechanisms of resistance[J]. International Journal of Cancer,2002,100(1):22-29.
[15] Kavanagh J J, Sill M W, Ramirez P T,etal. Phase II multicenter open-label study of karenitecin in previously treated epithelial ovarian and primary peritoneal cancer: a gynecologic oncology group study[J]. International Journal of Gynecological Cancer,2008,18(3):460-464.
[16] Tossell J A, Sahai N. Calculating the acidity of silanols and related oxyacids in aqueous solution[J]. Geochimica et Cosmochimica Acta,2000,64(24):4097-4113.
[17] Pooni P K, Showell G A. Silicon switches of marketed drugs[J]. Mini Reviews in Medicinal Chemistry,2006,6(10):1169-1177.
[18] 赵蓉,黄璐,耿海明.抗抑郁药盐酸文法拉辛的研究进展[J].广东化工,2009,36(12):98-99.
[19] Daiss J O, Burschka C, Mills J S,etal. Sila-venlafaxine, a sila-analogue of the serotonin/noradrenaline reuptake inhibitor venlafaxine: synthesis, crystal structure analysis, and pharmacological characterization[J]. Organometallics,2006,25(5):1188-1198.
[20] Warneck J B, Cheng F H M, Barnes M J,etal. Action ofR-sila-venlafaxine and reboxetine to antagonize cisplatin-induced acute and delayed emesis in the ferret[J]. Toxicology and Applied Pharmacology,2008,232(3):369-375.
[21] Daiβ J O, Burschka C, Mills J S,etal. Synthesis, crystal structure analysis, and pharmacological characterization of desmethoxy-sila-venlafaxine, a derivative of the serotonin/noradrenaline reuptake inhibitor sila-venlafaxine[J]. Journal of Organometallic Chemistry,2006,691(17):3589-3595.
[22] 孙弋阳,余辉.误服盐酸地芬尼多中毒1例[J]. Ment Retard Dev Disabil Res Rev,2002,8(2):61-65.
[24] Stoll C, Eltze M, Lambrecht G,etal. Functional characterization of muscarinic autoreceptors in rat and human neocortex[J]. J Neurochem,2009,110(3):837-847.
[25] 刘晓梅,廖仁安,谢庆兰.生物有机硅化合物研究新进展[J].有机化学,1998,18:397-402.
[26] Tacke R, Heinrich T, Bertermann R,etal. Sila-haloperidol: a silicon analogue of the Dopamine (D2) receptor antagonist haloperidol [J]. Organometallics,2004,23(19):4468-4477.
[27] Tacke R, Nguyen B, Burschka C,etal. Sila-trifluperidol, a silicon analogue of the Dopamine (D2) receptor antagonist trifluperidol: synthesis and pharmacological characterization[J]. Organometallics,2010,29(7):1652-1660.
[28] Tacke R, Popp F, Müller B,etal. Sila-haloperidol, a silicon analogue of the Dopamine(D2) receptor antagonist haloperidol: synthesis, pharmacological properties, and metabolic fate[J]. ChemMedChem,2007,3(1):152-164.
[29] Englebienne P, Hoonacker A V, Herst C V. The place of the bioisosteric sila-substitution in drug design[J]. Drug Design Reviews-Online,2005,2(6):467-483.
[30] Johansson T, Weidolf L, Popp F,etal.InVitrometabolism of haloperidol and sila-haloperidol: new metabolic pathways resulting from carbon/silicon exchange[J]. Drug Metabolism and Disposition,2010,38(1):73-83.
[31] 孙小星,郭红霞,梁英华,等.有机硅化合物应用进展[J].化工生产与技术,2007,14(2):51-54.
[32] Farag A T, Ibrahim H H. Developmental toxic effects of antifungal flusilazole administered by gavage to mice[J]. Birth Defects Research Part B: Developmental and Reproductive Toxicology,2007,80(1):12-17.
[33] 丛杉,沈德隆,孙娜波,等.广谱杀虫杀螨剂氟硅菊酯的合成综述[J].现代农药,2007,6(1):8-11.
[34] 缪康,束兆林,赵来成,等.醚菊酯和氟硅菊酯对秧田灰飞虱的防治效果[J].江苏农业科学,2009(6):176-177.
[35] Wilson S O, Tran N T, Franz A K. NMR and X-ray studies of hydrogen bonding for amide-containing silanediols[J]. Organometallics,2012,31(19):6715-6718.
[36] Sieburth S M N, Chen C A. Silanediol protease inhibitors: from conception to validation[J]. European Journal of Organic Chemistry,2006,2006(2):311-322.
[37] Juers D H, Kim J, Matthews B W,etal. Structural analysis of silanediols as transition-state-analogue inhibitors of the benchmark metalloprotease thermolysin[J]. Biochemistry,2005,44(50):16524-16528.
[38] Blunder M, Hurkes N, Spirk S,etal. Silanetriols asinvitroinhibitors for AChE[J]. Bioorganic & Medicinal Chemistry Letters,2011,21(1):363-365.
Progressonthe“SiliconSubstitution”ofDrugs
YE Tingting1,Wang Qiuyan2, SHEN Linghong1,CAO Dan2,WU Huili2,YIN Xiaopu2
(1. College of Life and Enviromental Sciences, Hangzhou Normal University, Hangzhou 310036, China; 2. Research Center of Biomedicine and Health, Hangzhou Normal University, Hangzhou 311121, China)
“Silicon substitution” of drugs is to synthesize silicon-containing analogs of the known drugs. Compared with the all-carbon counterparts, silicon-containing drugs have specific features, such as better curative effects, higher selectivity, less toxicity and side effects. Silicon substitution of drugs is a practical approach for drug design and innovation. This paper reviewed all kinds of silicon substitution of drugs.
silicon substitution; silicon-containing drug; drug design
2013-01-16
国家自然科学基金项目(21006018);杭州市科技局项目(20092113A03,20101131N03).
殷晓浦(1980—),男,助理研究员,博士,主要从事生物催化研究.E-mail:yinxp@hznu.edu.cn
10.3969/j.issn.1674-232X.2013.06.015
R914.2
A
1674-232X(2013)06-0555-07
