CmNn(m,n=1-10, 4≤m+n≤11)团簇的结构和稳定性研究
2013-10-28马志伟李宝兴顾娇娇
马志伟,李宝兴,顾娇娇
(杭州师范大学理学院,浙江 杭州 310036)
CmNn(m,n=1-10, 4≤m+n≤11)团簇的结构和稳定性研究
马志伟,李宝兴,顾娇娇
(杭州师范大学理学院,浙江 杭州 310036)
采用基于第一性原理的ADF程序,对锯齿型Nn(n=4-11)团簇掺杂质C原子后的稳定性以及它们的几何和电子结构等进行系统地研究.通过二级能量差计算得到原子总数为奇数的混合团簇中含有4个N原子的团簇较为稳定;而原子总数为偶数含有2个N原子的混合团簇较为稳定.文章还对纯N团簇锯齿形结构的成键特性进行了分析,解释了掺入多个杂质C原子后,C原子连线处出现结构被拉直的现象.
氮化碳团簇;几何结构;稳定性;轨道
0 引 言
氮是一种比较奇特的化学元素,通常的单质形态是氮气,无色无味无臭.N—N之间可以是单键、双键和三键,双键的强度高于单键强度2倍[1],叁键的强度又高于单键强度5倍之多.因此,以叁键存在的N2分子具有很高的稳定性,通常不易发生化学反应,而呈现化学惰性.碳是地球上最为丰富的化学元素,并以多种形式广泛存在于自然界中.常见的建筑材料钢铁中,碳元素是其主要成分之一.晶莹美丽、光彩夺目的金刚石是一种单质碳,它是自然界中最硬的物质.另一种由碳元素组成的用途广泛的石墨乌黑柔软,又是自然界中最软的矿石之一.随着C60富勒烯和纳米碳管等[2-4]的相继发现,人们对碳又有了新的认识,并因此开辟了一个全新的研究领域.

实验上对氮化碳团簇的研究也取得了进展.Garand等[6-7]通过高分辨率电子扫描成像得到C2nN-(n=1-3)基态均呈线性3∑-态;在实验上也验证了C2n+1N-稳定于C2nN-,并出现奇偶交替现象.
本文对Nn(n=4-11)团簇的结构、稳定性进行系统的研究,并将掺杂CmNn(m,n=1-10,4≤m+n≤11)团簇与纯氮团簇进行对比分析,在已有研究的基础上,发现碳掺杂时与氮原子的位置具有间隔分布的特点,并且,团簇的稳定性主要取决于N原子的个数.此外,文章还对电子轨道分布和成键角度进行分析,解释了纯氮团簇和掺杂团簇结构的成因.
1 计算方法
阿姆斯特丹密度泛函程序(Amsterdam density functional,简称ADF)采用了基于密度泛函理论的第一性原理方法[14],是目前国际上公认的用于团簇等研究的一种先进商业计算程序.在计算过程中,我们考虑了广义梯度近似(GGA),采用的是Becke-Perdew(B-P)交换关联泛函.
用该程序首先研究了Nn(n=4-11)团簇稳定结构,得到纯氮团簇为锯齿型线状构型,该结果与已有文献报道的完全吻合[15].然后按照排列组合的方式将纯氮团簇结构中的氮原子逐个替换为碳原子,直至所有的氮原子全部被替换,对这些替换得到的混合团簇及纯碳团簇的初始结构再次进行优化,最后得到各组分CmNn-m(m,n=1-10,4≤m+n≤11)团簇和线状纯碳团簇中最为稳定的结构.同时,还对文献报道中的环状纯碳团簇用ADF程序进行优化,并通过二级能量差和HOMO-LUMO能隙分析来研究它们的稳定性.
2 结果与讨论
2.1 CmN11-m团簇的结构与稳定性
对具有C2V对称性的锯齿型链状线性结构N11通过替代方式掺C杂质原子,得到1060个混合团簇CmN11-m(m=0-11)的初始结构.经结构优化后,得到各组分最为稳定的结构,如图1所示.当C原子数少于5个时,2个C原子不会出现在相邻位置上;当C原子数达到5个时,C原子与N原子交叉排列;当C原子数大于5个时,有多个C原子出现在相邻位置上,而且C原子连线处出现结构被拉直的现象;当C原子数超过9个时,结构几乎成为一条直线;随C原子数逐渐增加,首先被替代的是链中内部的氮原子,最后被替代的是链末端的氮原子.

浅色球代表N原子,深色球代表C原子图1 团簇CmN11-m(m=0-11)的结构图Fig. 1 The most stable structures of CmN11-m(m=0-11) clusters

a.团簇结合能的二级能量差;b.HOMO-LUMO能隙图2 团簇CmN11-m(m=0-11)Fig. 2 CmN11-m(m=0-11)clusters
通常根据团簇的二级能量差分值讨论它们的相对稳定性,二级能量差分值计算公式如下:
Δ2E=E(Cm-1N11-m+1)+E(Cm+1N11-m-1)-2E(CmN11-m),
(1)
其中,E(Cm-1N11-m+1)、E(Cm+1N11-m-1)和E(CmN11-m)分别为Cm-1N11-m+1、Cm+1N11-m-1和CmN11-m混合团簇的总能量.如果计算的Δ2E值为正,表明CmN11-m团簇比它相邻的Cm-1N11-m+1和Cm+1N11-m-1团簇更稳定.图2a给出了Δ2E与C原子数m的变化关系.当m>4时,这种稳定性表现出奇偶性,有奇数个C原子的团簇比有偶数个C原子的要稳定.图2b给出最高占据轨道(HOMO)与最低未占据轨道(LUMO)间的能隙Eg(单位eV)随m的变化规律.其中含有1个和7个C原子的混合团簇的能隙较大.另外,我们注意到,图1中11个原子的纯碳团簇为环型结构,计算表明它的结合能比替换得到的直线型结构(图1中未画出)C11的结合能要稳定0.74 eV.
2.2原子总数为奇数个的CmNn(m,n=1-9,4≤m+n≤9)团簇的结构及稳定性
同样利用原子替代的方式,可以得到原子总数为奇数个的混合团簇CmN9-m、CmN7-m以及CmN5-m的436个初始结构.图3给出了上述团簇经过优化后各组分最为稳定的结构.当C原子数少于N原子数时,2个C原子不会出现在相邻位置上;当C原子数比N原子数少1个时,C原子与N原子交叉排列;当C原子数多于N原子数时,有多个C原子出现在相邻位置上,且C原子连线处出现结构被拉直的现象;当N原子数小于或等于2个时,团簇结构几乎成为一条直线,但当团簇CmN5-m中N原子数为2个时,团簇成V字型结构.
图4分别给出了团簇CmN9-m、CmN7-m以及CmN5-m的二级能量差分值随m的变化关系,图4b、d、f为相应团簇的能隙随m的变化规律.对于团簇CmN9-m以及CmN7-m,图4a、c可以看出,含有奇数个C原子的团簇比有偶数个C原子的要稳定.结合图2和图4可以看到,对于总原子数相同的混合团簇,能隙最大的团簇组分相应Δ2E的值也最大,表现为具有较好的稳定性,如图2b中C7N4的能隙最大,图2a中C7N4的Δ2E值最小.另外,与C11不同的是,有9个和7个原子的纯碳团簇的直线型结构要比环型结构稳定(例如图4中C7比C7a稳定1.17 eV),但是含有5个原子纯碳团簇的环型结构不存在,因为它经过结构优化后,得到的仍为直线型结构.

浅色球代表N原子,深色球代表C原子图3 原子总数为奇数个的CmNn(m,n=1-9,4≤m+n≤9)团簇的结构图Fig. 3 The structures of odd number of CmNn(m,n=1-9,4≤m+n≤9)clusters

图4 奇数个原子团簇的二级能量差及HOMO-LUMO能隙Fig. 4 The second difference cluster energies and the energy gaps
2.3 原子总数为偶数个的CmNn(m,n=1-10,4≤m+n≤10)团簇的结构及稳定性
图5分别给出了通过原子替换得到的总原子数为偶数个的CmNn-m混合团簇(n≤10)经优化后的稳定结构.从图可以看到:除C3N5外,当C原子数与N原子数不相等时,结构规律类似于总原子数为奇数个的CmNn-m混合团簇(n≤11)的;当C原子数与N原子数相同时,有2个C原子相邻,其余C原子与N原子交叉排列;当其中含有1个或2个N原子时,团簇结构几乎成为一条直线,N原子位于结构末端,但当团簇CmN4-m中N原子数为1时,团簇内C原子间呈160.56°键角;当团簇内所有N原子被替换成C原子后,得到的纯C团簇为直线型结构.对于C10、C6、C4团簇,环型结构比直线型结构稳定,如图5所示,这与Leonid等[15]的结果一致.所不同的是,对于C8团簇,我们的计算结果表明,直线型结构却比环型结构要稳定,如图5中C8和C8a,前者比后者稳定1.43 eV.
另外,图5中结构N4是具有D∞h对称性的直线型结构,不同于具有C2h对称性的锯齿型线性结构.只要当4个N原子不在同一直线上时,其结构均分裂为2个N2分子.图5中混合团簇CmNn是通过C原子替换不在同一直线上的N原子经优化后得到的,可以看到,此时并没有N2分子分裂出来,且CN3和C3N出现锯齿型结构.此外,我们也将图5中结构N4内任意数目的N原子替换成C原子后进行优化,得到的结构均具有C∞v对称性,没有图中所示的各结构那样稳定.
图6a、c、e、g分别给出了团簇CmN10-m、CmN8-m、CmN6-m以及CmN4-m的二级能量差分值随m的变化关系,图6b、d、f、h为相应团簇的能隙随m的变化规律.从二级能量差分图可以看到,除团簇C6N4外,总原子数为偶数个的混合团簇CmNn(n≤10)的稳定性出现奇偶交替现象,即有偶数个C原子的团簇比有奇数个C原子的要稳定;与奇数个总原子数的混合团簇相同的是,能隙最大的团簇组分相应Δ2E的值也最大,表现为稳定性较好.
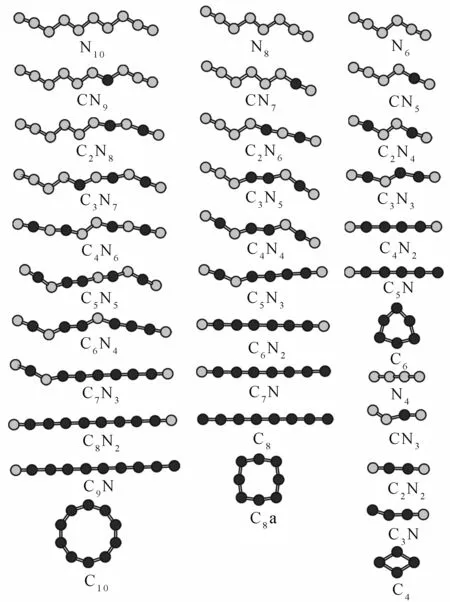
浅色球代表N原子,深色球代表C原子图5 原子总数为偶数个的CmNn(m,n=1-10,4≤m+n≤10)团簇的结构图Fig. 5 The structures of even number of CmNn (m,n=1-10,4≤m+n≤10)clusters
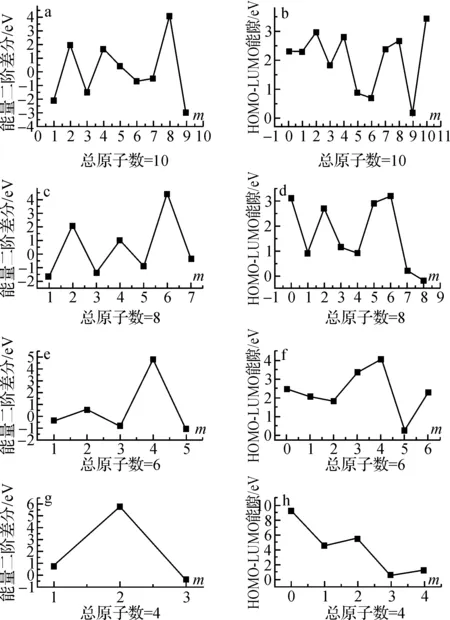
图6 偶数个原子团簇的二级能量差及HOMO-LUMO能隙Fig. 6 The second difference cluster energies and the energy gaps
2.4 纯N团簇及CmNn(m,n=1-11,4≤m+n≤11)团簇结构的成因分析

图7 团簇N9的两种结构图Fig. 7 The structures of N9cluster

a.HOMO(-5.806 eV);b.HOMO-1(-6.602 eV);c.HOMO-2(-7.537 eV);d.HOMO-3(-7.682 eV).括号中为 所处能级的能量图8 团簇N9的电子云图Fig. 8 The electronic of N9 cluster
前面讨论过纯N团簇均呈线性的锯齿型链状结构,为了探究其原因,我们对N原子团簇的成键轨道进行了分析.由于σ轨道上的电子所占据的能量要比π轨道上的电子占据的能量低,电子云图上显示的σ-π轨道是分离的,也正因此,与稳定性和化学反应活性密切相关的前线轨道(包括HOMO和LUMO)通常为π轨道.本文中,除N5和N10外,其余Nn(4≤n≤11)团簇的前线轨道均为π轨道.同时,由于N元素的2s和2p轨道能级差值小,s-p轨道混杂作用显著[16-17].以N9为例说明,图7a是N9团簇的稳定结构,而图7b不能稳定存在.图8是优化后N9团簇(几何结构如图7a,原子标号参考图7)电子云图,蓝色小球代表N原子,呈不规则蓝色和红色的片状部分代表电子云,其中a为HOMO轨道,b、c、d能级依次降低,图8b所在能级二重简并,能差仅为0.08 eV,它们电子云图相同.从成键角度来看,电子云相对于结构平面前后对称分布,如图8a(电子云俯视图,相对于图7a结构).很明显,原子2、原子3以及端位的原子8、原子9形成单电子π键,原子6、原子7分别与相邻的原子4、原子5形成π键;图8b(次轨道的电子云正面图),中心原子1分别与相邻原子2和原子3经s-p轨道混杂形成两个σ键,标号为2、3、4、5的原子的组键类似原子1,而对于边缘处原子(标号为6、7、8、9),由于电荷转移导致原子内形成能量更低的单电子π键,正如图8c(下一低能级轨道的电子云俯视图)所示,Mulliken电荷布居也显示处于6、7位置的原子电荷分布为正,电子转移到其它原子上.图8d是图8c下一低能级轨道的电子云正面图,其中原子2和原子4,原子3和原子5分别形成一个σ键,原子8和原子9各形成一个单电子π键.根据上述分析,N原子间形成两个σ键一个π键的组合,导致整个N原子链呈锯齿形状,理论上键角为90°,但由于s-p轨道混杂,轨道之间的相互作用,使得键角处于104.28°~113.25°之间.
图7b所示结构不能稳定存在,根据成键分析,我们可以解释其中原因.从图8a、c、d中,可以看到处于端位的原子8和原子9的轨道为3个单电子π键,链中并没有多余的电子与之形成σ键,所以N原子团簇中处于端位的原子与相邻原子键角接近180°,如图7a所示,而不是图7b的锯齿形状.C元素比N元素最外层少1个电子,将N团簇中掺入C杂质原子后,C原子与相邻原子形成π键,所以出现了团簇“被拉直”的现象.
3 小 结
结合以上分析发现,CmNn(m,n=1-10,4≤m+n≤11)团簇结构和稳定性具有如下特点:
1.当C原子数比N原子数少时,除团簇C3N5外,2个C原子不会出现在相邻位置上;当C原子数比N原子数少1个时,C原子与N原子交叉排列;当C原子数等于N原子数时,有2个C原子相邻,其余C原子与N原子交叉排列;当C原子数比N原子数多时,有多个C原子出现在相邻位置上,且C原子连线处出现结构被拉直的现象.
2.团簇的二级能量差分曲线表明,原子总数为奇数个的混合团簇,含有4个N原子的混合团簇稳定性较好;原子总数为偶数个的混合团簇,含有2个N原子的混合团簇稳定性较好;总原子数相同时能隙最大的团簇对应的稳定性也较好.
3.纯N团簇内部原子的s-p轨道混杂,轨道的相互作用导致内部原子键角处于104.28°~113.25°之间,且N—N间形成两个σ键一个π键呈锯齿链状结构,而多个C原子相邻时键角接近180°,则是C杂质原子间形成π键的结果.
[1] 史启祯.无机化学与化学分析[M].2版.北京:高等教育出版社,2005:356.
[2] Kroto H W, Heath J R, O’Brien S C,etal. C60: Buckminsterfullerene[J]. Nature,1985,318(14):162-163.
[3] Iijima S. Helical microtubules of graphitic carbon [J]. Nature,1991,354(7):56-58.
[4] Li W Z, Xie S S, Qian L X,etal. Large-scale synthesis of aligned carbon nanotubes[J]. Science,1996,274(5293):1701-1703.
[5] Liu A Y, Cohen M L.Prediction of new low compressibility solids [J]. Science,1989,245(4920):841-842.
[6] Garand E, Yacovich T I, Neumark D M. Slow photoelectron velocity-map imaging spectroscopy of C2N-, C4N-, and C6N-[J]. J Chem Phys,2009,130(6):064304-064310.
[7] Wang Chunru, Huang Rongbin, Liu Zhaoyang,etal. Laser generation and ab initio studies of CnN-clusters [J]. Chem Phys Lett,1995,237(5):463-467.
[8] Li Niu, Wang Xuanzhang, Zhu Jiaqi,etal. First-principles studies of the vibrational properties of amorphous carbon nitrides [J]. Chin Phys B,2013,22(1):017101.
[9] Angelou G C, Church R P, Stancliffe R J,etal. Thermohaline mixing and its role in the evolution of carbon and nitrogen abundances in globular cluster red giants: the test case of messier 3 [J]. The Astrophysical Journal,2011,728:79-90.
[10] Jiang Zhenyi, Xu Xiaohong, Wu Haishun,etal. Theoretical study of structures and stabilities of CmN2(m=1-14) ions [J]. Int J Mass Spectrom,2003,230(1):33-39.


[13] Mogren M M, El-Azhary A A, Alkiali W Z.A G3 study of the structure of carbon-nitrogen nanoclusters [J]. J Phys Chem A,2010,114(46):12258-12268.
[14] Lenthe E, Baerends E J.Optimized Slater-type basis sets for the elements 1-118 [J].J Comput Chem,2003,24(9):1142-1156.
[15] Thompson M D, Bledson T M, Strout D L. Dissociation barriers for odd-numbered acyclic nitrogen molecules N9and N11[J]. J Phys Chem A,2002,106(29):6880-6882.
[16] Belau L, Wheeler S E, Ticknor B W,etal. Ionization thresholds of small carbon clusters: tunable VUV experiments and theory [J]. J AM Chem Soc,2007,129(33):10229-10243.
[17] 周公度.结构和物性:化学原理的应用[M].2版.北京:高等教育出版社,2009:44-51.
TheStructureandStabilityofCmNn(m,n=1-10,4≤m+n≤11)Clusters
MA Zhiwei, LI Baoxing, GU Jiaojiao
(College of Science, Hangzhou Normal University, Hangzhou 310036, China)
This paper systematically studied the stability, geometric and electronic structure of the zigzag Nn(n=4-11) clusters doped with carbon atoms by using the Amsterdam Density Functional (ADF) program based on the First Principle. By calculating the second difference of cluster energies, the results show that the clusters containing 4 nitride atoms are more stable in the hybrid clusters with odd total number atoms, and the clusters containing 2 nitride atoms are more stable in the hybrid clusters with even total number atoms. The paper also analyzed the bond character of zigzag pure nitride clusters, and explained the phenomenon that carbon atoms join was straightened after the clusters doped with multiple carbon atoms.
carbon nitride clusters; geometric construction; stability; orbital
2012-10-07
浙江省自然科学基金项目(Y6100098).
李宝兴(1960—),男,教授,主要从事团簇研究.E-mail: phybxli@aliyun.com
10.3969/j.issn.1674-232X.2013.04.015
O469
A
1674-232X(2013)04-0359-06
