样品前处理技术在气相色谱分析中的应用进展
2013-10-22严矿林林丽琼郑夏汐肖小华曹玉娟
严矿林, 林丽琼, 郑夏汐, 肖小华*, 曹玉娟
(1.中山大学化学与化学工程学院,广东广州 510275;2.华南师范大学化学与环境学院,广东广州 510006)
气相色谱法(GC)对分析样品的要求较高,大多数样品必须经过前处理后才能进行分析。复杂基体样品前处理的步骤往往繁琐耗时、易引起误差,已成为制约分析效率和准确度提升的关键环节,成为分析化学的瓶颈问题,因此样品前处理技术研究越来越受到分析工作者的重视。从2009.1.1-2013.5.6这一时期主要样品前处理技术在GC中应用的相关中英文文献发表情况(见图1)来看,吹扫捕集(P&T)、固相萃取(SPE)、固相微萃取(SPME)、液相微萃取(LPME)和热解吸(TD)技术得到了大量应用,以微波辅助萃取(MAE)、超声波辅助萃取(UAE)为代表的各种场辅助萃取技术因适合固体样品前处理而日益受到重视。本文综述了2009-2013年几种主要样品前处理技术,包括 P&T、SPE、SPME、LPME、MAE、UAE 等在GC分析中的应用。
1 P&T技术
P&T技术快速准确、富集效率高且无需有机溶剂,已成为一种通用、高效的气相色谱/质谱(GC/MS)联用分析样品的前处理技术,适合萃取沸点低于200℃、溶解度小于2%(v/v)的挥发性或半挥发性有机物气体样品。随着商用吹扫捕集仪器的广泛使用,P&T法在食品、环境等领域的挥发性有机物分析检测中起着越来越重要的作用。但气体吹扫时在液体样品中容易形成泡沫,使仪器超载;且吹出的水蒸气不利于下一步吸附捕集,给非极性气相色谱柱的分离带来困难。Ruiz-Bevia等[1]用改进的P&T法分析了水中的三卤甲烷类化合物。他们先将25 mL水样放入吹扫管中,然后用氦气等惰性气体吹扫样品,三卤甲烷分析物在捕集管上用液体CO2冷却吸附,待样品吹扫捕集完成后,将捕集管热解吸,分析物采用GC/MS分析。实验证明该方法快速、灵敏、高效。此外,该方法也已成功应用于蜂蜜[2]、污水[3]等样品以及蓝藻水华样品[4]中特征化合物的分析中,检出限可低至0.1 μg/L。

图1 2009.1.1-2013.5.6期间主要样品前处理技术在气相色谱法中的应用Fig.1 Sample preparation techniques used in GC analysis during 2009.1.1-2013.5.6
2 SPE技术
SPE技术操作简便快速、富集倍数高且易于实现自动化或与其他分析仪器联用,因此逐渐取代液液萃取法成为液体样品处理优先考虑的方法。近20年来,SPE与GC、GC/MS等在线联用分析技术在食品、环境、生物和医药等复杂基体痕量目标物分析中得到了广泛应用[5],目前GC分析中有超过三分之一的样品采用SPE技术进行样品前处理。Muller等[6]采用 SPE-GC/MS法分析了自来水和饮用水中超痕量的21种氟苯甲酸,检出限低至6~44 ng/L。
SPE装置通常以填充柱、整体柱形式存在,即在管状或饼状的中空容器(两端开口)中填充颗粒固定相,或直接在该容器中合成具有大量可流通空隙的整体柱固相萃取材料,其中萃取材料是SPE性能指标的核心。一些新型材料如功能化硅胶、石墨烯、碳纳米管等因良好的吸附性能而在SPE等分离富集中具有良好的应用前景。以多壁碳纳米管为吸附剂,采用SPE-GC/MS分析环境水样中的多环芳烃时,检出限可达2.0~8.5 ng/L[7]。采用磺化石墨烯片为吸附剂,μ-SPE-GC/MS分析水样中多环芳烃类化合物(PAHs)的检出限也可达到0.8~3.9 μg/L[8]。而以吩噻嗪键合硅胶为吸附剂,SPE-GC/MS方法分析环境水样中硝基苯类化合物的检出限更是低至0.06~0.3 ng/mL[9]。除了碳材料外,具有良好构效预定性、特异识别性和广泛实用性的分子印迹材料在复杂基体中选择性分离和富集结构类似的痕量分析物中显示出了很好的应用潜力[10,11]。采用分子印迹材料作为固相萃取的吸附剂,可以选择性地萃取PAHs,污水样品中16种PAHs的检出限为 5.2 ~12.6 ng/L[10]。
3 MSPD技术
与SPE技术相比,MSPD技术所需的样品和有机溶剂用量少,并避免了样品均化、沉淀、离心、乳化和浓缩等环节可能造成的目标物损失,操作简单快速,特别适合固体、半固体和黏性样品的前处理。自1989年该技术问世以来,MSPD与GC或GC/MS相结合,在环境、医药卫生、食品等方面得到了成功应用[12,13],并有取代传统的索氏抽提等前处理技术的趋势。基质分散的程度以及分散剂的性质是影响该方法萃取性能的关键因素。除了C18等亲脂性分散剂、弗罗里硅土等正相分散剂以及硅藻土等中性分散剂之外,多壁碳纳米管和金属有机骨架化合物(MOFs)等新材料由于巨大的比表面积成为MSPD理想的分散/吸附材料。Su等[14]以多壁碳纳米管为分散剂,建立了奶油样品中的多种雌激素的MSPDGC/MS分析方法,雌二醇等8种雌激素的检出限为 0.2 ~1.3 μg/kg,定量限 0.8 ~4.5 μg/kg。而以对苯二酸酯铝配合物(MIL-53)和1,3,5-苯三羧酸三乙酯铜配合物(BTC)等MOFs材料作为MSPD吸附剂萃取虎尾草中莠灭净等7种农药残留后,其GC-MS分析方法的检出限可低至0.02~0.07 μg/g[15]。与传统的C18吸附剂相比,MOFs等新材料的萃取性能具有明显的优势。
当分散剂与目标分子的相互作用过强时,通过一些外场的强化作用可以提高目标分子的萃取效率。如采用超声波对基质和分散剂混合物进行处理,有利于提高分散剂的萃取效果。与PLE或MAE等技术联用,可以充分利用高温高压或微波作用下溶剂的洗脱性能,在保持目标物萃取选择性的同时提高其萃取效率。通过MSPD与SPME能够有效去除复杂基体中杂质的干扰,结合GC-ECD,可以高选择、高灵敏地测定生物样品中的有机磷农药或多溴联苯醚类残留[16]。
4 SPME技术
固相微萃取集采样、萃取、富集和进样于一体,具有耗时少、效率高、操作简单等优点,是一种无溶剂或少溶剂的样品前处理技术。与固相萃取技术类似,固相微萃取技术也是气相色谱分析中主要的样品前处理技术。它可以以纤维针式 SPME、管内SPME、固态搅拌棒萃取等多种形式处理复杂基体中的目标物,在食品、环境、天然产物等领域的挥发性有机物分析中应用广泛[17-19]。离子液体因对有机物和无机物都具有良好的溶解性且其结构可调的优点,作为萃取介质在SPME中有良好的应用潜力。Ho等[20]以离子液体[C4MIM][PF6]涂覆 SPME 纤维,结合GC/MS建立了垃圾渗滤液中氯酚的分析方法,其中五氯苯酚的检出限为0.008 μg/L。而Amini等[21]将离子液体键合到硅胶上作为SPME介质,建立了汽油中甲基叔丁基醚的顶空SPME-GC/FID分析方法,检出限为0.1 μg/L。此外,碳纳米管、金属有机骨架化合物等新材料也作为SPME的萃取介质在食品[22]、环境[23]等领域得到成功应用。
由于搅拌棒的体积比探针纤维大,因此搅拌棒SPME的萃取容量相比纤维式SPME的容量更大。Liu等[24]发展了一种便捷、可自动搅拌的自组装中空纤维搅拌棒微萃取装置。萃取用的中空纤维搅拌棒固定在磁力搅拌子上,同时将萃取溶剂(接受相)放入中空纤维中并将纤维两端封口,从而有效地减少了萃取时有机溶剂挥发带来的影响,提高了分析方法的精密度和回收率。采用该方法分析了生物样品中的7种合成类固醇,检出限可达0.1 ng/mL。
5 LPME技术
传统的液-液萃取法有机溶剂消耗多、环境污染较重。以此为基础,Jeannot等[25]于1996年提出了LPME技术,它集萃取、净化、浓缩和预分离为一体,具有萃取效率高、溶剂消耗少的优点,可以方便地与GC、LC等色谱及各种检测器进行连接,实现样品前处理与分析测定的在线联用。对于一些沸点比较低的化合物,如食品、环境样品中的有机氯农药残留[26]、短链脂肪酸[27]或者药品中的溶剂残留[28],可以直接通过单液滴微萃取或分散液液微萃取(DLLME)之后,采用GC或GC/MS分析。而对于一些极性较大、沸点较高的化合物,如生物胺[29]、长链脂肪酸[30]等,可以在萃取时对目标物进行衍生化,然后再结合 GC或 GC/MS分析。如 Almeida等[29]建立了一种 DLLME-衍生化-GC/MS的方法,用于同时测定啤酒中的18种生物胺。该方法采用乙腈为分散剂,甲苯为萃取剂,氯甲酸异丁酯作为衍生化试剂,样品经萃取并衍生化后采用GC/MS分析,所有生物胺的检出限均低于2.9 μg/L。如果将DLLME技术与其他前处理技术如固相萃取等联合使用,可进一步提高分析物的富集倍数和萃取效率。Zhao等[31]以多壁碳纳米管作为SPE吸附剂先净化环境水样,然后进一步采用DLLME富集,结合GC/MS分析了水中的酰胺类除草剂,目标物的富集倍数为6593~7873倍,检出限低至 0.002~0.006 μg/L。
离子液体因具有良好的溶解性和化学物理性质可调节的优点,适合作为单液滴微萃取的萃取液滴[32,33]。Chisvert 等[33]采用 1-甲基-3-己基咪唑六氟磷酸盐离子液体作为萃取相,结合热解吸-GC/MS分析了水样中的10种氯苯类化合物。为此,他们设计了一个双层玻璃萃取管,其中20 mm长的内管用于盛放萃取用的离子液体,187 mm长的普通热解吸管作为外管,在内管的两端填入玻璃棉。实验证实采用这一萃取管可以有效地防止解吸进样时离子液体进入气相色谱柱中。
6 场辅助样品前处理技术
虽然如前所述的基于两相或多相分离的吹扫捕集技术、固/液相微萃取技术、基质固相分散萃取技术等样品前处理技术得到了快速发展,碳纳米管、石墨烯、分子印迹聚合物、金属有机骨架化合物、离子液体等新型分离介质在气体和液体样品前处理中得到了良好的应用,但对于固体或半固体样品而言,分析物首先需要从固体基质中有效、快速地转移到相应的萃取溶剂中,然后才能开展进一步的分离富集和分析检测。通过引入适当的相、膜和场等外部作用力给体系增加能量和降低体系的熵值,从而促进固体样品中的分析物与溶剂之间的相对迁移和物料平衡是提高这一过程效率的有效手段。近年来,采用热、声、电、磁、力和微波场等外场作用强化样品处理过程中传热和传质过程,加快样品的处理速度、提高处理效率的前处理技术得到了较快的发展。
6.1 UAE 技术
UAE技术利用超声波的空化作用和热作用加快分析物从固体基质中溶解出来以提高其萃取效率。它作为一种成熟的前处理技术在环境、食品等领域已有广泛应用[34]。由于UAE技术的选择性不高,在复杂基体中痕量目标物分析中经常与其他样品前处理技术配合使用,加快样品前处理的速度。如采用UAE与顶空单液滴微萃取(HS-SDME)结合分析土壤中的有机磷农药残留[35];与DLLME结合分析水样中的邻苯二甲酸酯类化合物[36];或先采用超声波辅助热水萃取,然后再通过单液滴微萃取进一步净化,结合GC/MS分析土壤中的氯苯类化合物[37]。
在天然产物挥发油的萃取分析中,可以采用超声波雾化萃取(UNE)的方式先将挥发油从固体样品中萃取到水等溶剂中,然后再用十七烷等溶剂为HS-SDME的萃取相富集挥发油组分,用GC/MS进行分析测定。Wang等[38]采用该方法分析了中药小茴香中的挥发油组分。实验证明,相比于传统的搅拌萃取-HS-SDME联用技术以及单独的 UAE或UNE技术,UNE-HS-SDME技术可以使小茴香中的伞花素、萜品烯等挥发油组分的富集效率和灵敏度得到显著提高。
6.2 MAE 技术
微波辅助萃取技术是在微波场中通过微波作用强化传热和传质的一种样品萃取技术。与浸提、溶剂回流、UAE等技术相比,MAE不仅萃取效率高、能耗小、操作费用少,且符合环境保护要求,广泛应用于中草药、香料、食品和化工等诸多领域[39-41]。
在微波场作用下对目标物进行 SPE[42]、SPME[43]、MSPD[44]、LPME[45]或者 P&T 萃取[46],可以实现微波技术与多种萃取技术之间的优势互补,减少溶剂消耗、加快处理过程、提高萃取效率,在环境污染物、食品危害因子、生物样品分析等方面都得到了成功应用。Zhou等[47]研制了微波辅助索氏固相萃取(MA-SSE)装置(图2a):索氏萃取器中填有玻璃滤片,玻璃滤片上方可承载吸附剂,构成固相萃取结构,从而在微波辅助索氏萃取的同时完成净化。以此为基础,他们发展了集萃取、净化为一体的MA-SSE联用新技术,建立了西洋参中8种有机磷及氨基甲酸酯农药残留的MA-SSE-GC/MS分析方法。随后,他们[48]又研制了微波超声辅助固液固分散萃取(HF-SLSDE)装置(图2b),通过样品和弗罗里硅土等分散剂组成两个固相,萃取溶剂为液相,发展了集萃取、净化为一体的HF-SLSDE联用新技术;并建立了烟草中13种有机氯农药残留的HF-SLSDE-GC/ECD分析方法,取得了令人满意的分析结果。
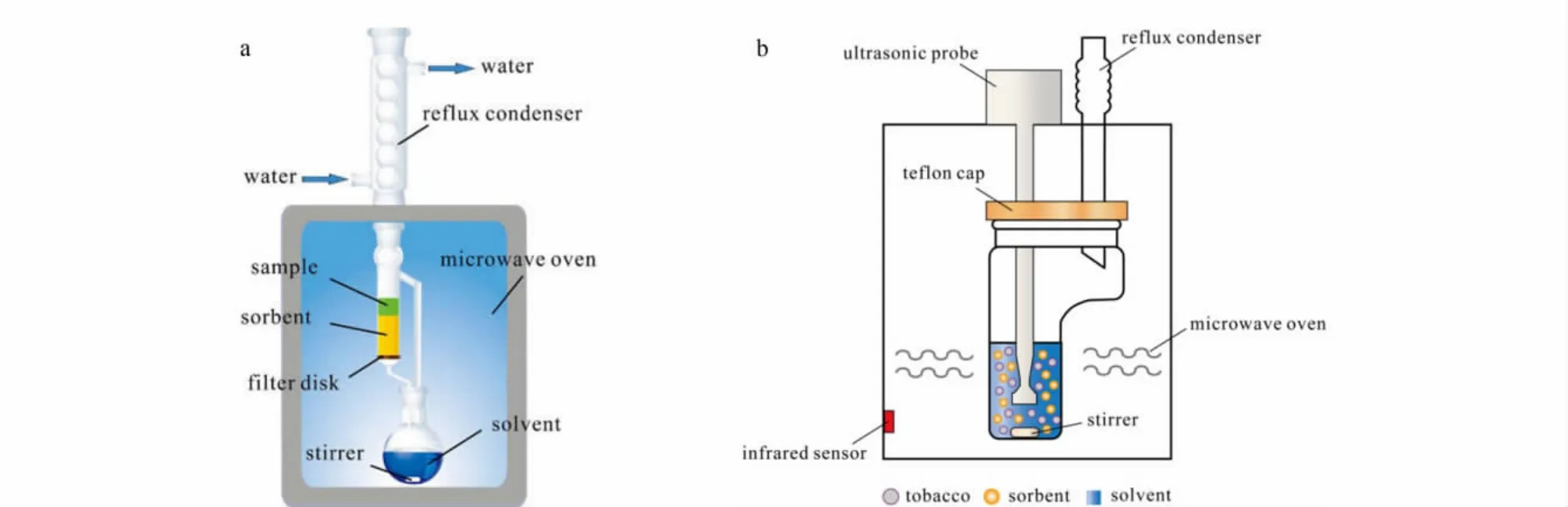
图2 (a)微波辅助索氏固相萃取[47]和(b)微波超声辅助固液固分散萃取[48]装置示意图Fig.2 The schematic diagrams of(a)MA-SSE[47] and(b)HF-SLSDE[48] apparatus
6.3 超临界流体萃取(SFE)技术
SFE是以超临界状态下的流体为萃取溶剂分离萃取混合物的过程。超临界流体具有类似于气体的较强穿透力和类似于液体的较大密度和溶解度,具有良好的溶剂特性,它克服了传统的索氏萃取费时费力、回收率低、重现性差、污染严重等弊端,使样品的萃取过程更加快速简便,特别是消除了有机溶剂对人体和环境的危害。CO2气体的临界温度(31.06℃)和临界压力(7.39 MPa)较低,是常用的超临界流体,适合于萃取非极性和中等极性的物质,且萃取出的分析物通常可以直接采用GC或GC/MS分析[49]。
对于一些极性较大的分析物,可以在CO2超临界流体中加入适量的夹带剂如甲醇、乙醇等调节其极性;或结合其他前处理技术进行分析。Yang等[50]构建了一种 SFE-原位衍生-HS-SPME萃取装置用于与GC/MS在线联用分析化妆品中的苯甲酸酯类防腐剂和多酚类抗氧化剂。分析物采用CO2超临界流体萃取后,直接用双甲基硅烷三氟乙酰胺(BSTFA)和三甲基氯硅烷收集并衍生化,GC/MS分析,8种防腐剂和氧化剂的检出限为0.5~8.3 μg/g。采用类似的装置和方法,Liu等[51]分析了沉积物中的全氟羧酸,其检出限为0.39~0.54 ng/g。
6.4 PLE 技术
PLE技术是在压力场和温度场(热场)的综合作用下,通过加压实现在高于正常溶剂沸点的温度下进行快速萃取的一种前处理技术,适用于固体和半固体样品的前处理。尽管该技术1995年才提出,但由于具有萃取时间短、萃取效率高、溶剂消耗量少、操作模式多样化以及操作过程自动化等诸多优点,受到国外分析化学界的极大关注,在环境、药物、食品和聚合物工业等领域得到广泛应用[52],并迅速被接受为美国EPA SW-846方法的3545A号标准。
以水为溶剂的加压热水萃取法(PHWE)绿色环保、萃取效率较高,是一种具有良好应用前景的样品前处理技术。采用加压热水萃取法萃取,进一步衍生化并结合SPME等净化技术和GC/MS等分析方法,可以实现复杂基体中极性较大的目标物的快速高效分析,如Ramirez等[53]采用PHWE法萃取室内尘埃中的对羟基苯甲酸乙酯类化合物,分析物经乙酰化衍生后用搅拌棒固相微萃取(SBSE)并采用TD-GC/MS分析,检出限为1.0~2.1 ng/g,定量限为3.3~8.5 ng/g。Llpo等[54]采用 PHWE 结合 HSSPME萃取,GC/MS分析了污水污泥中的9种亚硝胺,检出限也低至 0.15 μg/kg。
7 其他样品前处理技术
膜分离技术具有装置简单、操作程序方便、无需有机溶剂处理、可与各种分析仪器直接连接、易于实现自动化操作和在线操作等优点,已经成功应用到包括各种气体、液体、固体样品等多种样品的基体分离和预富集,可以进行挥发性、半挥发性甚至不挥发性物质的分离与浓缩。与顶空、吸附、低温和微捕集等分离技术的联用使膜分离技术成为当前各种分析样品制备的主要方法和热点研究领域之一。自20世纪70年代发展了膜分离技术与气相色谱及质谱的接口以来,相继出现了膜引进质谱、膜-GC/MS、膜-微捕集/质谱、膜萃取-GC等联用技术,膜在挥发性有机物的分离和在GC和MS分析样品制备中的应用研究越来越多,发展也越来越快[55,56]。
热解吸技术通常与前面介绍的SPE、SPME、P&T、膜分离等样品前处理技术配合使用,主要用于从固体吸附剂上将目标物解吸下来,热解吸与GC或GC/MS联用具有广泛的应用范围。如顶空吸附萃取-热解吸-GC/MS分析环境样品中的有机锡类化合物[57]、热解吸-二维 GC-飞行时间质谱(TOF/MS)分析人类组织中的挥发性化合物(VOCs)[58]等。
此外,凝胶色谱(GPC)[59]、QuEChERS[60]等样品前处理技术与GC、GC/MS技术结合使用在环境、食品等领域均有良好的应用。
8 结论及展望
样品前处理是色谱分析中耗时最多、最容易引起误差的关键步骤,直接影响色谱分析结果。近年来有关色谱分析的前处理技术受到了分析工作者的广泛重视,一些新的前处理技术不断出现,一些新型材料也快速地应用到样品前处理技术中,多种样品前处理联用技术及样品前处理-气相色谱在线联用技术逐渐成为分析化学的研究热点。样品前处理技术及其与气相色谱联用技术的不断发展,必将大幅度提高色谱分析方法的准确度、精密度和分析速度,推进色谱技术在食品、生物、环境、医药等诸多领域中的应用。
[1]Ruiz-Bevia F,Fernandez-Torres M J,Blasco-Alemany M P.Anal Chim Acta,2009,632:304
[2]Soria A C,Martinez-Castro I,Sanz J.J Chromatogr A,2009,1216:3300
[3]Barco-Bonilla N,Plaza-Bolanos P,Fernandez-Moreno J L,et al.Anal Bioanal Chem,2011,400:3537
[4]Deng X,Liang G,Chen J,et al.J Chromatogr A,2011,1218:3791
[5]Jimenez J J.Anal Chim Acta,2013,770:94
[6]Muller K,Seubert A.J Chromatogr A,2012,1260:9
[7]Ma J,Xiao R,Li J,et al.J Chromatogr A,2010,1217:5462
[8]Zhang H,Low W P,Lee H K.J Chromatogr A,2012,1233:16
[9]Peng X T,Zhao X,Feng Y Q.J Chromatogr A,2011,1218:9314
[10]Santos M G,Vitor R V,Andrade F L,et al.J Chromatogr B,2012,909:70
[11]Song X L,Li J H,Xu S F,et al.Talanta,2012,99:75
[12]Labadie P,Alliot F,Boourges C,et al.Anal Chim Acta,2010,675:97
[13]Garcia-Fodriguez D,Cela-Torrijos R,Lorenzo-Ferreira R A,et al.Food Chem,2012,135:259
[14]Su R,Wang X H,Xu X,et al.J Chromatogr A,2011,1218:5047
[15]Aquino A,Ferreira J A,Navickiene S,et al.J AOAC Int,2012,95:1338
[16]Moliner-Martinez Y,Campins-Falco P,Molins-Legua C,et al.J Chromatogr A,2009,1216:6741
[17]Kenessov B N,Koziel J A,Grotenhuis T,et al.Anal Chim Acta,2010,674:32
[18]de Morais P,Stoichev T,Basto M C P,et al.Anal Bioanal Chem,2011,399:2531
[19]Riu-Aumatell M,Vargas L,Vichi S,et al.Food Chem,2011,129:557
[20]Ho T T,Chen C Y,Li Z G,et al.Anal Chim Acta,2012,712:72
[21]Amini R,Rouhollahi A,Adibi M,et al.J Chromatogr A,2011,1218:130
[22]Sarafraz-Yazdi A,Abbasian M,Amiri A.Food Chem,2012,131:698
[23]Chen X F,Zang H,Wang X,et al.Analyst,2012,137:5411
[24]Liu W,Zhang L,Fan L B,et al.J Chromatogr A,2012,1233:1
[25]Jeannot M A,Cantwell F F.Anal Chem,1996,68(13):2236
[26]Cortada C,Vidal L,Tejada S,et al.Anal Chim Acta,2009,638:29
[27]Li Y,Xiong Y Q,Liang Q Y,et al.J Chromatogr A,2010,1217:3561
[28]Yu Y J,Chen B,Shen C,et al.J Chromatogr A,2010,1217:5158
[29]Almeida C,Femandes J O,Cunha S C.Food Control,2012,25:380
[30]Pusvaskiene E,Januskevic B,Prichodko A,et al.Chromatographia,2009,69:271
[31]Zhao R S,Diao C P,Chen Q F,et al.J Sep Sci,2009,32:1069
[32]Aguilera-Herrador E,Lucena R,Cardenas S,et al.J Chromatogr A,2009,1216:5580
[33]Chisvert A,Roman I P,Vidal L,et al.J Chromatogr A,2009,1216:1290
[34]Sanchez-Brunete C,Miguel E,Albero B,et al.J Chromatogr A,2010,1217:7024
[35]Salemi A,Rasoolzadeh R,Nejad M M,et al.Anal Chim Acta,2013,769:121
[36]Yan H Y,Liu B M,Du J J,et al.Analyst,2010,135:2585
[37]Wang L,Wang L L,Chen J,et al.J Chromatogr A,2012,1256:9
[38]Wang L,Wang Z M,Zhang H H,et al.Anal Chim Acta,2009,647:72
[39]Bowden J A,Colosi D M,Stutts W L,et al.Anal Chem,2009,81:6725
[40]Coscolla C,Castillo M,Pastor A,et al.Anal Chim Acta,2011,693:72
[41]Liu R L,Zhang J,Mou Z L,et al.Analyst,2012,137:5135
[42]Azzouz A,Ballesteros E.J Chromatogr B,2012,891/892:12
[43]Ponnusamy V K,Jen J F.J Chromatogr A,2011,1218:6861
[44]Huang M C,Chen H C,Fu S C,et al.Food Chem,2013,138:227
[45]Gholivand M B,Abolghasemi M M,Piryaei M,et al.Food Chem,2013,138:251
[46]Deng X W,Xie P,Qi M,et al.J Chromatogr A,2012,1219:75
[47]Zhou T,Xiao X H,Li G K.Anal Chem,2012,84:5816
[48]Zhou T,Xiao X H,Li G K.Anal Chem,2012,84:420
[49]Chikushi H,Fujii Y,Toda K.J Chromatogr A,2012,1256:267
[50]Yang T J,Tsai F J,Chen C Y,et al.Anal Chim Acta,2010,668:188
[51]Liu W L,Hwang B H,Li Z G,et al.J Chromatogr A,2011,1218:7857
[52]Aragon M,Marce R M,Borrull F.Talanta,2012,101:473
[53]Ramirez N,Marce R M,Borrull F.J Chromatogr A,2011,1218:6226
[54]Llpo A,Borrull F,Pocurull E.Talanta,2012,88:284
[55]Shi X Z,Song S Q,Sun A L,et al.Analyst,2012,137:437
[56]Posada-Ureta O,Olivares M,Navarro P,et al.J Chromatogr A,2012,1227:38
[57]Cacho J I,Campillo N,Vinas P,et al.J Chromatogr A,2013,1279:1
[58]Stadler S,Stefanuto P H,Brokl M,et al.Anal Chem,2013,85(S1):998
[59]Xu X M,Yu S,Li R,et al.Food Chem,2012,135:161
[60]Hou X,Han M,Dai X H,et al.Food Chem,2013,138:1198
