Zr0.5Ti0.5O2的添加对超临界裂解RP-3催化剂性能的影响
2013-10-18秦莉晓李雄健王健礼李象远陈耀强
焦 毅 秦莉晓 李雄健 王 佳 王健礼,*朱 权 李象远 陈耀强
(1四川大学化学学院,绿色化学与技术教育部重点实验室,成都 610064;2四川大学化工学院,成都 610064)
1 引言
由于高超声速飞行器具有较高的热载荷,超燃冲压发动机的再生冷却近几年被提上了发动机设计的日程.单靠碳氢燃料的物理热沉,难以实现对高超音速条件下工作的发动机实现有效冷却,因此在燃料进入燃烧室前实现热裂解或催化裂解并产生较大的化学热沉,是实现发动机再生冷却设计的重要环节.1-4由热力学可以知道,燃料裂解若形成乙烯、丙烯和丁烯等不饱和烃和氢气,反应吸热量大,且烯烃分子量越小吸收的热量越多,化学热沉越高,此种裂解气的点火性能好;而形成甲烷、乙烷和丙烷等饱和烃,化学热沉低,同时点火性能差.因此,希望吸热燃料在裂解反应中按照生成小分子烯烃和氢气的途径进行,生成尽可能多的乙烯、丙烯、丁烯等低碳烯烃,从而获得较高的热沉.利用催化剂的选择性,可显著地减少烷烃的生成,促进燃料选择性裂解,生成更多的氢气和低碳烯烃.5-7
吸热性碳氢燃料催化裂解是一个复杂的平行反应体系,裂解反应的同时,其他一些异构化、氢转移、双烯缩合等副反应也在进行,其中对裂解多产烯烃最不利的反应就是氢转移反应,氢转移反应的结果是消耗了烯烃产生了更多的烷烃和芳烃,因此要提高烯烃的含量就必须严格控制氢转移反应的进行.8,9C―C键的断裂需在催化剂的强酸中心上进行,而氢转移反应可在不同的酸中心上进行,所以提高催化剂表面强酸中心的比例,可提高C―C键的断裂反应与氢转移反应的速度之比,从而提高烯烃选择性.10-13对于负载型催化剂,载体对催化剂的催化性能有着重要的影响.14,15目前,最常用的催化剂载体有Al2O3、SiO2、活性炭和分子筛等.本课题组长期从事催化载体的研究和应用,耐高温大比表面积的氧化铈氧化铝催化剂用于吸热性碳氢燃料裂解反应,已取得了比较好的催化效果,该催化材料高温热稳定性、催化活性都比较理想,但是没有表现出更好的选择性,经研究发现是由于该载体材料的表面酸性较弱.由于ZrO2、TiO2复合氧化物具有更大的比表面积,更好的热稳定性和机械强度以及更强的表面酸碱性,16-18ZrO2-TiO2复合氧化物近年来已引起人们极大的关注,已广泛用于乙酸正丁酯的酯化反应,19水杨酸的光致氧化和Cr(VI)的光致还原,20柴油车尾气中颗粒物的氧化去除,21NOx的储存还原法(NSR)催化剂的抗S中毒载体22等.还有研究23表明用Al2O3和ZrO2-TiO2作为双组分载体对水汽变换反应有较好的催化性能.
Zr0.5Ti0.5O2载体材料具有比较强的表面酸性和相对集中的酸性中心密度,能够催化燃油以正碳离子机理进行裂化反应,有利于裂解反应的进行,这样却将伴随着很多裂解副反应的进行,氢转移反应增加,容易积碳堵塞催化剂管道,因此采用Zr0.5Ti0.5O2载体添加到CeO2-Al2O3(CA)基催化剂中,以降低弱酸中心密度,提高强酸中心比例,控制氢转移反应,来改善乙烯等烯烃的选择性.本文首先着眼于催化剂管式涂层技术的实现,在此基础上采用航空煤油RP-3进行了热裂解和催化裂解的对比研究,评价了催化剂的催化效果.
2 实验部分
2.1 载体材料及催化剂的制备
采用共沉淀法制备了Zr0.5Ti0.5O2复合氧化物载体材料.24,25分别以硝酸锆(分析纯,山东鱼台清达精细化工厂)和硫酸氧钛(化学纯,丹东化学试剂厂)为前驱体,按一定的化学计量比溶于水中配成一定浓度的溶液,用一定比例的氨水和碳酸铵的混合物作为沉淀剂进行沉淀,所得沉淀经水浴陈化、洗涤、干燥,600°C焙烧3 h.同样方法制得CeO2-Al2O3(Ce:Al摩尔比为1:1)复合氧化物载体材料.将得到的CeO2-Al2O3载体材料,以氯铂酸为前驱体,采用等水孔体积浸渍法将Pt负载于CeO2-Al2O3材料上(Pt 0.50%(w)).120°C烘干,550°C焙烧2 h,得到Pt/CA催化剂粉料.
将上述所得的Pt/CA催化剂粉料分成两份,再加入Ce(NO3)3·6H2O(化学纯,四川乐山市五通桥东风化工厂)和La(NO3)3·6H2O(化学纯,上海跃龙有色金属有限公司),ZrOCO3(分析纯,成都科龙化学试剂厂)作为助剂前驱体,在其中一份中加入10%(w)的Zr0.5Ti0.5O2载体材料,并与水用球磨机混合制浆,涂覆在处理过后的不锈钢管内壁(上载量为0.2 g/70 cm),然后在120°C烘干,再于550°C焙烧2 h,即可得涂层催化剂,记做Cat2,另一组Pt/CA催化剂粉料未加入Zr0.5Ti0.5O2载体的催化剂记做Cat1.
2.2 催化剂评价

图1 实验装置示意图Fig.1 Schematic diagram of apparatus
采用本实验室组装的吸热燃料超临界裂解装置评价催化剂的催化活性,超临界裂解装置由进料系统、计量系统、温控系统、反应系统、压力系统、分析系统组成.如图1所示,反应管是70 cm长,内径2 mm的304不锈钢管.在压力3.5 MPa(RP-3超临界压力2.390 MPa,超临界温度372.4°C),燃料RP-3流量1.0 g·s-1的工况下进行实验,高压计量泵连续流动,两点式电加热的方式给反应系统提供反应所需温度.裂解气相产物经气液分离罐分离后通过六通阀直接进入气相色谱仪(GC2000 III上海计算技术研究所),选用HP-Al/S毛细管分离柱(安捷伦科技有限公司,50 m×0.53 mm)和氢火焰离子化检测器(FID)检测有机小分子烃类,用实验室自己装配的2 m填充柱(固定相,5A分子筛)和热导检测器(TCD)检测氢气等无机小分子,回收的液体采用气相色谱与质谱联用(GC/MS)分析.并在实验中对几个温度产气率、热沉做了测试.热沉采用加热时消耗的电功率进行测定.实验中以管道本身的电阻为加热元件,采用电加热的方法加热流体.加热功率W乘以热效率η即是流体的吸热量.对加热功率的测量只需测出电压U和电流I.吸热量除以质量流量就可得出热沉,如式(1)所示.

式中:Qm为热沉(MJ·kg-1);G为质量流量(kg·s-1);W为加热功率(kW);η为热效率;U为电压(V);I为电流(A).
2.3 催化剂表征
催化剂的织构性能用QUADRASORB型比表面测定仪(美国康塔公司)测定.样品首先在300°C下抽真空处理1 h,用氮气作为吸附质,在液氮温度(-196 °C)下进行测量.
材料的结构采用DX-2500型旋转阳极X射线衍射仪(中国丹东方圆仪器有限公司)进行分析.采用Cu Kα辐射,石墨单色器,Ni滤波片,电压为40 kV,管电流为25 mA,扫描速率为0.03(°)·s-1,扫描范围(2θ)为10°-80°.
采用Tecnai G2F20 S-TWIN型高倍透射电镜(美国FEI公司)观察催化剂的微形貌特征.
采用TP-5076型TPD动态吸附仪(天津先权公司)测试材料的表面酸性.样品的用量为100 mg,首先样品在20 mL·min-1的氮气流吹扫下以8°C·min-1升温速率加热升温到400°C,恒温45 min后降温,然后切换为NH3(2%)+N2(98%)混合气吸附60 min,流速为20 mL·min-1,待降至室温后,以8 °C·min-1的升温速率升温至950°C,TCD检测.
3 结果与讨论
3.1 Zr0.5Ti0.5O2载体材料的添加对裂解反应结果的影响
3.1.1 产气率
图2给出了热裂解与催化裂解产气率比较图,由图可知,加入催化剂后各个温度产气率均有所提高,750°C时Cat2产气率已达到54.5%.600、650、700°C时Cat1较热裂解产气率分别提高了2.8倍、38.5%、64.1%,Cat2分别提高了4.0倍、64%、95.2%.而Cat2在Cat1的基础上又提高了30%左右.为达到相同的产气率,催化剂所需的反应温度要低于热裂解所需的温度,催化剂可以有效降低裂解反应的起始温度,催化剂的加入明显地增加了裂解的反应深度,使得反应物分子的活化能降低,反应速率增加,26,27提高了各种小分子烃类产物的生成量.
3.1.2 气相分布和烯烷比
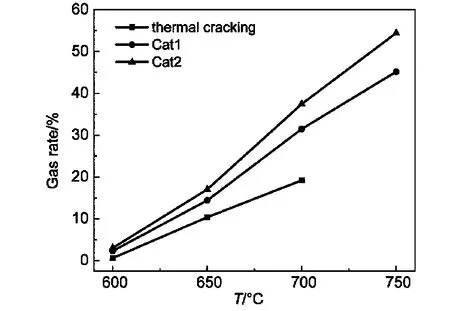
图2 热裂解与催化裂解的产气率Fig.2 Gas rates of thermal cracking and catalytic cracking
表1给出了每个反应温度,催化裂解与热裂解气相产物分布规律,由上表可知RP-3裂解气相产物主要由氢气、甲烷、乙烷、乙烯、丙烷、丙烯、C4组成,并随着温度的升高和催化剂的加入各组分含量在不断变化.热裂解氢气含量随温度升高,逐渐增加,C1、C2随温度的升高呈先增大后减少,C3、C4则先减少后增加,裂解反应是一个吸热反应,同时也是体系内分子数增加的反应.根据热力学原理,温度升高有利于提高裂解率,使得C1、C2含量增加,当达到一定温度时,由于体系内分子的混乱度增加,分子间互相碰撞的几率增大,导致小分子C1、C2的二次反应几率增加,因此体系内C3、C4出现了先减少后增加的趋势.28催化裂解与热裂解相比氢气含量变化不大,说明催化剂主要不是脱氢作用,甲烷含量减少,乙烯、丙烯含量增加,这主要是由于加入催化剂后,裂解反应按照自由基和C+离子两种机理29同时进行,改变了裂解反应的历程和反应速度.催化剂的加入不仅提高了催化剂的活性,也提高了催化剂的烯烃选择性,热裂解600°C由于产气量较少,只有很少一部分裂解,裂解过程中产生烯烃量大于烷烃,但是随着温度的升高,烯烃的摩尔含量先减少后增多.C―C键的断裂需在催化剂的强酸中心上进行,而氢转移反应可在不同的酸中心上进行,提高催化剂表面强酸中心的比例,可提高C―C键的断裂反应与氢转移反应的速度之比,从而提高反应的烯烃生成量.Cat1主要以弱酸和中强酸为主,催化活性提高,烯烃选择性提高不明显.Cat2中加入了部分Zr0.5Ti0.5O2载体,烯烃摩尔比例增加明显,各个温度点都比Cat1催化剂烯烃收率高出几乎两倍,可能是由于增强了催化剂的表面酸性,提高了强酸中心密度,抑制了氢转移生成烷烃、芳烃的反应,有利于生成更多的烯烃.下文用NH3-TPD结果对这一现象进行详细解释.
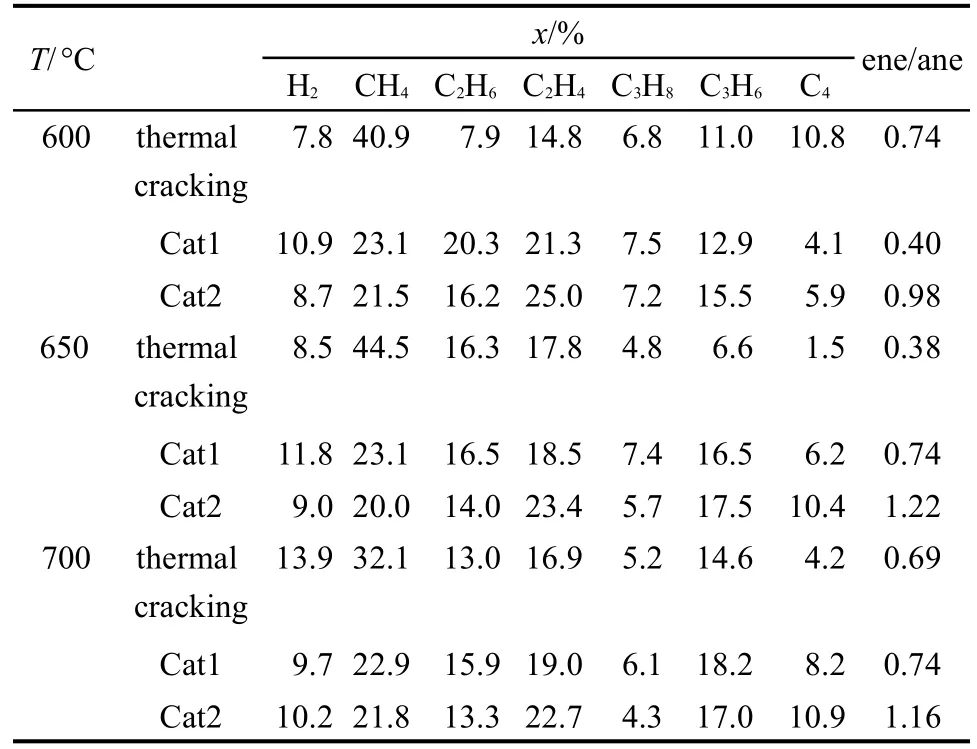
表1 各温度点下气相产物分布Table 1 Gas phase product distribution of thermal cracking and catalytic cracking
3.1.3 热沉结果
由图3可以看出,热沉随着温度升高明显增加,且Cat1和Cat2催化裂解反应的热沉值均高于热裂解.700°C热裂解由于管道积碳严重,停止反应.650°C时,Cat2较热裂解热沉提高了0.55 MJ·kg-1,750°C时Cat2的热沉已达到3.83 MJ·kg-1.600、650、700°C各温度点下Cat1较热裂解分别提高了2.3%、11.2%、12%,Cat2较热裂解分别提高了11.4%、29.8%、18.8%.600、650、700、750 °C时Cat2比Cat1热沉分别提高了8.7%、15.9%、6.1%、4.2%.催化剂的加入使得裂解反应向有利于生成小分子烯烃的方向进行,烯烃的生成量Cat2>Cat1>thermal cracking,与热沉大小一致,裂解生成烯烃的反应是吸热反应,能够产生更高的热沉.
3.2 催化剂的织构性能
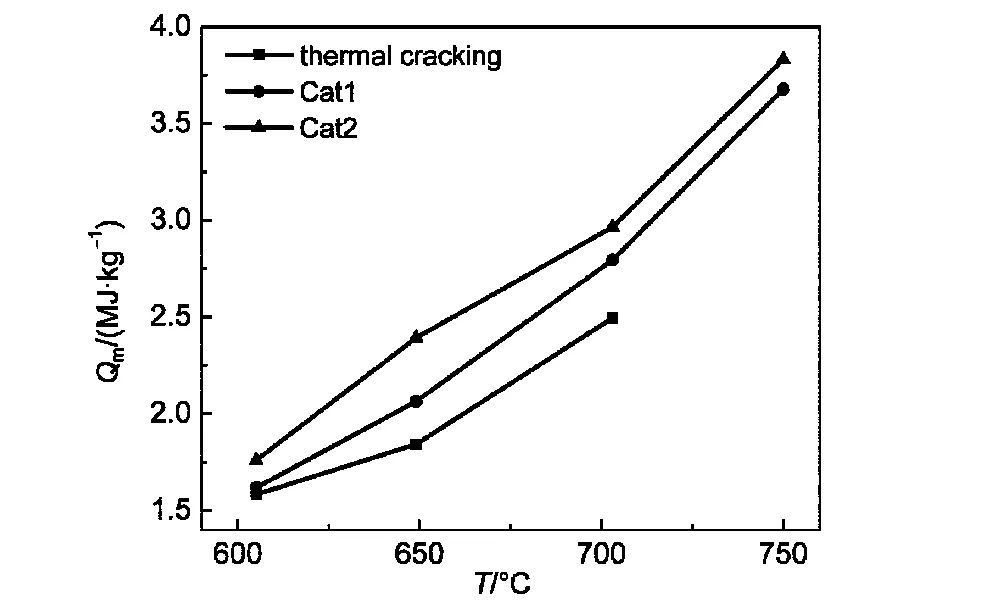
图3 热裂解与催化裂解的热沉(Q m)分布图Fig.3 Heat sink(Q m)distribution of thermal cracking and catalytic cracking

表2 两组催化剂和Zr0.5Ti0.5O2载体材料的织构性能Table 2 Textural performance of two catalysts and Zr0.5Ti0.5O2support material
表2列出了两组催化剂和Zr0.5Ti0.5O2载体材料的比表面积、孔容和平均孔径结果.可以看出,Zr0.5Ti0.5O2载体材料的添加使得催化剂的比表面积降低和平均孔径减小,催化剂的比表面积对催化剂的催化活性有一定的影响,孔大小和孔形状对催化剂的选择性和活性也有影响,对于催化裂解多产烯烃反应小孔有利于小分子烃类的通过,而积碳则容易在大孔内部堆积,阻止催化反应的进行.30
3.3 两组催化剂和Zr0.5Ti0.5O2载体材料的孔径分布和N2吸附-脱附等温线
图4为两组催化剂和Zr0.5Ti0.5O2载体材料的孔径分布图,BJH公式算得催化剂的孔径分布(图4(a))可知,两组催化剂孔径大多处于2-14 nm之间,介孔居多,最可几孔径为6 nm左右,且Cat2和Zr0.5Ti0.5O2载体均存在双峰.孔径较小的可能是材料孔壁中少量固体颗粒堆积而成的无序间隙孔道;孔径较大的为模板剂填充形成的有序介孔.31Cat2孔径在2和6 nm处集中分布,而Zr0.5Ti0.5O2载体则以2-3 nm的小孔为主,表明正是由于Zr0.5Ti0.5O2载体的添加使得Cat2孔向小孔移动,也有可能是由于Zr0.5Ti0.5O2载体进入了CA催化剂的孔中,双孔则有利于催化裂解反应中不同大小活化分子在不同大小的孔中进行反应.32由图4(b)催化剂的N2吸附-脱附等温线图可知,三组样品均表现为典型的具有介孔材料(2-50 nm)特征的IV型吸附等温线,并且由于毛细管凝聚现象,所有样品均有明显的滞后环,33属于典型的H2型,表明样品的孔形状为狭缝型和瓶型.34吸附支曲线在相对压力(p/p0)为0.7-0.9处吸附量迅速增大,这说明样品具有较大的平均孔径,相对集中的孔径分布,且以2-50 nm的介孔为主,这与孔径分布结果一致.同时这种滞后环表明样品的孔大小形状不完全均一,由滞后环可判断在p/p0接近1时,吸附脱附曲线才达到平衡.
3.4 XRD表征
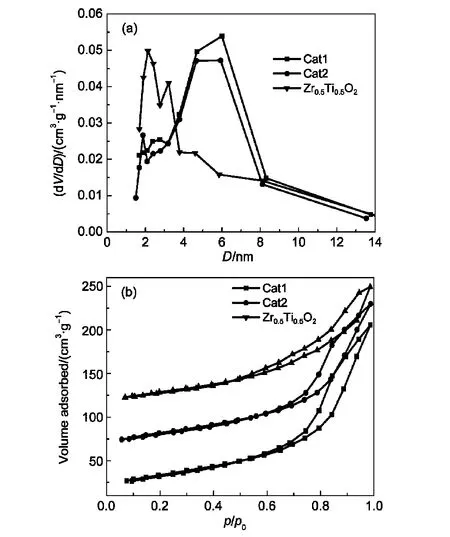
图4 两组催化剂和Zr0.5Ti0.5O2载体材料的孔径分布(a)和N2吸脱附曲线(b)Fig.4 Pore diameter distributions(a)and N2adsorptiondesorption isotherms(b)of two catalysts and Zr0.5Ti0.5O2support material

图5 不同催化剂的XRD谱图Fig.5 XRD patterns of different catalysts
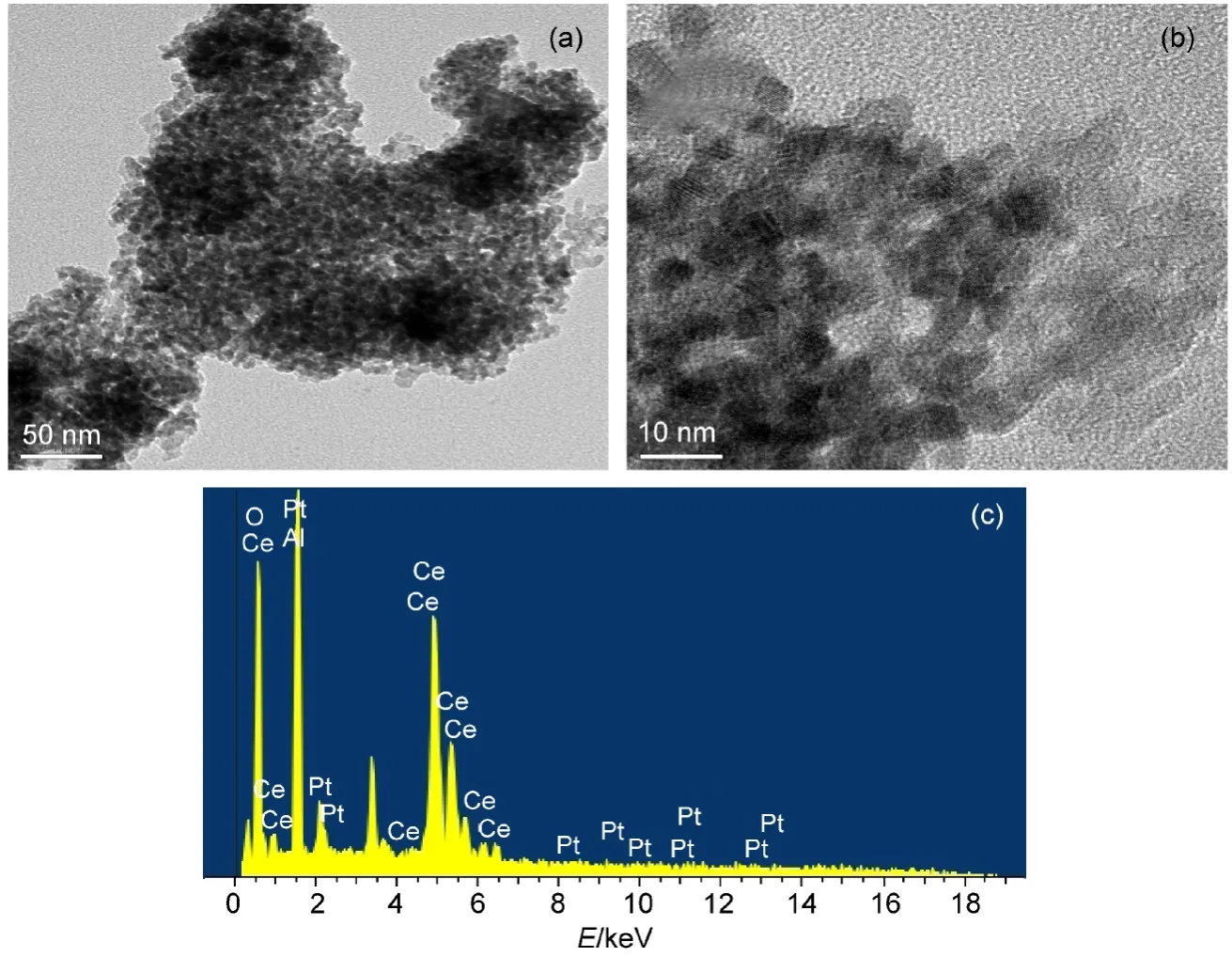
图6 Pt/CA催化剂的TEM图(a,b)和能量色散X射线谱(EDX)(c)图Fig.6 TEM(a,b)and energy dispersive X-ray spectroscopy(EDX)(c)of Pt/CAcatalyst
图5为两组催化剂和Zr0.5Ti0.5O2载体材料XRD图谱.由图可见Cat1和Cat2的衍射谱峰基本相同,在28.8°、32.9°、47.4°、56.4°、76.7°等处有谱峰出现,可归属为立方萤石结构CeO2晶体.35在67.1°处的衍射峰为γ-Al2O3的谱峰.而在600°C焙烧3 h的Zr0.5Ti0.5O2载体材料只有部分晶化,在24.8°、30.7°和53.6°处有衍射峰,归属为ZrTiO4谱峰,并没有出现ZrO2和TiO2衍射峰,说明ZrO2的加入阻碍了TiO2晶型转变,这与文献36,37报道一致.XRD结果表明两组催化剂主要以CeO2立方萤石结构和γ-Al2O3晶相存在.Cat2中并未出现ZrTiO4谱峰,说明加入Cat2中的Zr0.5Ti0.5O2载体形成的ZrTiO4高度分散于CeO2立方萤石晶体和γ-Al2O3晶相上,形成了更细小的晶粒,XRD未检测出来,而添加到Cat2中的Zr0.5Ti0.5O2载体只有很少一部分晶化,研究表明Zr0.5Ti0.5O2载体经650°C焙烧后出现了比较完整的ZrTiO4晶相特征衍射峰,800°C焙烧后晶化趋于完成,1000°C焙烧时晶相基本不再变化.38,39复合氧化物晶化后,比表面积减低,表面酸性减弱,都会减弱催化剂的催化活性,Zr0.5Ti0.5O2载体添加到CA基催化剂中高度分散形成了更细小的晶粒,阻碍了高温下裂解反应过程中ZrTiO4晶型的完整,有利于催化反应的进行.Cat1、Cat2谱图中都未出现Pt的衍射峰,说明Pt是以无定形态存在或者高度分散于载体中,40有利于催化剂活性的提高.
3.5 TEM表征
图6是Cat1(Pt/CA)的微观形貌图.如图6所示,在低分辨率TEM图(a)和高分辨率HRTEM图(b)中均没有发现Pt粒子,Pt并没有团聚现象,但经能量色散X射线谱(EDX)对催化剂一小部分区域进行元素扫描,图(c)中催化剂不仅出现了C、Al和O,还出现了Pt元素,结合HRTEM和EDX说明Pt是高度分散于载体材料中,这与XRD分析结果基本一致,活性组分高度分散有利于催化剂催化活性的提高和催化剂的利用效率.
3.6 NH3-TPD表征
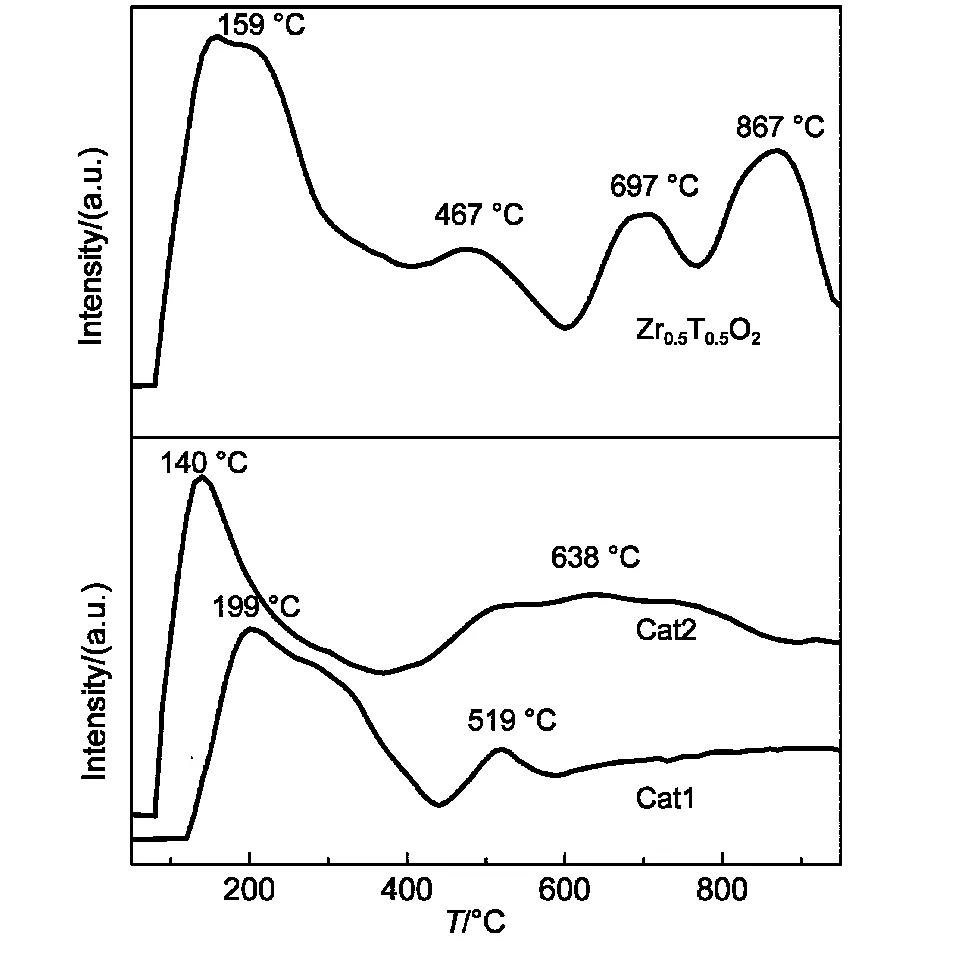
图7 不同催化剂的NH3-TPD结果Fig.7 NH3-TPD results for different catalysts

表3 催化剂的酸量分布Table 3 Acidity of catalysts
图7是两组催化剂和Zr0.5Ti0.5O2载体的NH3-TPD图,两组催化剂表面吸附的NH3呈连续脱附状态,表明两组催化剂表面酸中心强度呈非均一化连续分布且表面的酸性位种类较多.由图中可以看出Cat1、Cat2都在低温(100-400°C)区有一个强的脱附峰,归属为弱酸和中强酸的脱附峰,400-800°C的高温区脱附峰归属为强酸的脱附峰,且Cat2脱附峰面积明显比Cat1要大得多.Zr0.5Ti0.5O2载体则具有更宽的脱附范围和更高温度的脱附峰,Zr0.5Ti0.5O2载体材料的添加增加了CA基催化剂的强酸脱附峰面积,提高了CA基催化剂的表面酸性和提高了强酸酸中心比例.由表3可知催化剂的总酸量Cat2>Cat1,且Cat2的强酸酸量较大,达到0.082 mmol·g-1是Cat1的4.0倍.Zr0.5Ti0.5O2载体的添加,使催化剂的强酸酸量增加.结合NH3-TPD图和酸量分布则可得到Cat2具有较强的表面酸性和集中的强酸中心密度.文献11,12报道C―C键的断裂需在催化剂的强酸中心上进行,而其最主要的竞争反应氢转移反应则在不同的酸性位都可以进行,氢转移反应主要生成烷烃和大分子芳烃,容易积碳,提高催化剂酸性的同时要增加强酸酸性中心比例来提高烯烃的选择性.Cat1主要以弱酸和中强酸为主,且中强酸的酸量较大,Cat2的催化剂样品主要以中强酸和强酸中心为主.Cat2中加入10%的Zr0.5Ti0.5O2载体明显增强了催化剂的酸性和提高了催化剂的强酸中心密度,使得Cat2催化剂在高温区的峰面积增加,低温区的峰向更低的温度区域移动,强酸中心密度更集中,使得裂解反应的选择性提高.
4 结论
考察了CA催化剂和CA催化剂中添加部分Zr0.5Ti0.5O2载体对煤油超临界裂解反应的影响.催化剂能够明显地降低催化裂解反应的温度,600°C Cat1催化剂的产气率是该温度点下热裂解的2.8倍,加入10%的Zr0.5Ti0.5O2的CA基催化剂较热裂解时提高4.0倍.650°C时,Cat2催化剂较热裂解热沉提高了0.55 MJ·kg-1.BET结果表明,加入Zr0.5Ti0.5O2的催化剂的孔径出现小孔,提高了催化裂解乙烯的选择性.NH3-TPD结果表明,加入Zr0.5Ti0.5O2载体的CA基催化剂,Cat2的强酸酸量增加了4.0倍,催化剂表现出更强的强酸中心密度和更强的表面酸性,有利于裂解反应的进行.
(2)Kay,I.W.;Peschke,W.T.J.Propul.Power 1992,18(2),507.
(3)Lander,H.R.;Nixon,A.C.ACS Div.Pet.Chem.1987,32,504.
(4)Lander,H.;Nixon,A.C.J.Aircraft.1971,8,200.doi:10.2514/3.44255
(5)Wickham,D.T.;Atria,J.V.;Engel,J.R.;Hitch,B.D.;Karpuk,M.E.;Striebich,R.ACS Div.Pet.Chem.1998,43,428.
(6)Buchanan,J.S.Catal.Today 2000,55,207.doi:10.1016/S0920-5861(99)00248-5
(7)Edwards,T.ACS Div.Pet.Chem.2000,45,436.
(8)Wickham,D.T.;Engel,J.R.;Hitch,B.D.J.Propul.Power 2001,17,1253.doi:10.2514/2.5872
(9)Wickham,D.T.Methods for Suppression of Filamentous Coke Formation.US Patent 6482311 B1,2002,2003-03-07
(10)Fărcasiu,D.;Le,K.H.Catal.Commun 2001,2,5.
(11)Zhao,G.L.;Teng,J.W.;Xie,Z.S tud.Surf.Sci.Catal.2007,1307.
(12)Dimitris,K.L.;David,T.A.Ind.Eng.Chem.Res.1992,31,45.doi:10.1021/ie00001a007
(13)Tsai,T.C.;Kung,H.Y.;Yu,S.T.Appl.Catal.A 1989,50,1.doi:10.1016/S0166-9834(00)80820-9
(14)Benjaram,M.R.;Biswajit,C.;Panagiotis,G.S.Appl.Catal.A:Gen.2001,211,19.doi:10.1016/S0926-860X(00)00834-6
(15)Benjaram,M.R.;Ataullah,K.Catal.Rev.2005,47,257.doi:10.1081/CR-200057488
(16)Ma,Z.Y.;Xu,R.;Yang,C.Acta Ph ys.-Chim.Sin.2004,20,1221.[马中义,徐 润,杨 成.物理化学学报,2004,20,1221.]doi:10.3866/PKU.WHXB20041011
(17)Naoki,T.;Akihiko,S.;Ichiro,H.Appl.Catal.B-Environ.2007,72,187.doi:10.1016/j.apcatb.2006.10.014
(18)Han,C.H.;Liu,B.H.;Zhang,H.L.Acta Phys.-Chim.Sin.2006,22,993.[韩承辉,刘炳华,张惠良.物理化学学报,2006,22,993.]doi:10.1016/S1872-1508(06)60044-2
(19)Das,D.;Mishra,H.K.;Parida,K.M.J.Mol.Catal.A:Chem.2002,189,271.doi:10.1016/S1381-1169(02)00363-1
(20)Colón,G.;Hidalgo,M.C.;Navo,J.A.Appl.Catal.A:Gen.2002,23,185.
(21)Oi-Uchisawa,J.;Wang,S.D.;Nanba,T.Appl.Catal.B-Environ.2003,203,207.
(22)Matsumoto,S.;Ikeda,Y.;Suzuki,H.Appl.Catal.B-Environ.2000,25,115.doi:10.1016/S0926-3373(99)00124-1
(23)Laniecki,I.M.;MaLecka,G.M.;Domka,F.Appl.Catal.A:Gen.2000,196,293.doi:10.1016/S0926-860X(99)00480-9
(24)Mao,D.S.;Lu,G.Z.;Chen,Q.L.Chin.J.Catal.2004,25,501.[毛东森,卢冠忠,陈庆龄.催化学报,2004,25,501.]
(25)Yu,Y.;Lin,T.;Zhang,L.J.;Guo,J.X.;Gong,M.C.;Chen,Y.Q.J.Inorg.Meter.2003,23,71.[喻 瑶,林 涛,张丽娟,郭家秀,龚茂初,陈耀强.无机材料学报,2003,23,71.]
(26)Fu,X.C.;Shen,W.X.;Yao,T.Y.;Hou,W.H.Physical Chemistry;Higher Education Press:Beijing,2006;pp 191-197.[傅献彩,沈文霞,姚天扬,侯文华.物理化学.北京:高等教育出版社,2006:191-197.]
(27)Jiao,Y.;Li,J.;Wang,J.B.;Wang,J.L.;Zhu,Q.;Chen,Y.Q.;Li,X.Y.Acta Phys.-Chim.S in.2011,27,1061.[焦 毅,李 军,王静波,王健礼,朱 权,陈耀强,李象远.物理化学学报,2011,27,1061.]doi:10.3866/PKU.WHXB20110437
(28)Wang,Z.W.;Zhang,X.W.;Mi,Z.T.;Hao,W.H.Petrochem.Tech nol.2005,6,518.[王占卫,张香文,米镇涛,郝伟华.石油化工,2005,6,518.]
(29)Fărcasiu,D.;Vargas,W.;Lee,K.H.Catal.Commun 2003,4,63.
(30)Babitz,S.M.;Williams,J.T.;Miller,R.Q.Appl.Catal.A:Gen.1999,179,71.doi:10.1016/S0926-860X(98)00301-9
(31)Nie,R.F.;Wang,J.H.;Fei,J.H.;Hou,Z.Y.;Zheng,X.M.Chin.J.Catal.2011,32,379.[聂仁峰,王军华,费金华,侯昭胤,郑小明.催化学报,2011,32,379.]
(32)Sobel,D.R.;Spadaccini,L.J.J.Eng.Gas.Turb.Power 1997,119,344.doi:10.1115/1.2815581
(33)Zhang,L.J.;Dong,W.P.;Guo,J.X.;Yuan,S.H.;Zhang,L.;Gong,M.C.;Chen,Y.Q.Acta P hys.-Chim.Sin.2007,23,1738.[张丽娟,董文萍,郭家秀,袁书华,张 磊,龚茂初,陈耀强.物理化学学报,2007,23,1738.]doi:10.3866/PKU.WHXB20071116
(34)Leofanti,G.;Padovan,M.;Tozzola,G.;Venturelli,B.Catal.Today 1998,41,207.doi:10.1016/S0920-5861(98)00050-9
(35)Deraz,N.M.Ceram.Trans.2012,38,747.
(36)Lin,T.;Li,W.;Gong,M.C.;Yu,Y.;Du,B.;Chen,Y.Q.Acta P hys.-Chim.Sin.2007,23,1851.[林 涛,李 伟,龚茂初,喻 瑶,杜 波,陈耀强.物理化学学报,2007,23,1851.]doi:10.1016/S1872-1508(07)60089-8
(37)Lin,T.;Zhang,Q.L.;Li,W.;Gong,M.C.;Xing,Y.X.;Chen,Y.Q.Acta P hys.-Chim.Sin.2008,24,1127.[林 涛,张秋林,李 伟,龚茂初,幸怡汛,陈耀强.物理化学学报,2008,24,1127.]doi:10.1016/S1872-1508(08)60046-7
(38)Mao,D.S.;Lu,G.Z.;Chen,Q.L.;Xie,Z.K.;Zhang,Y.X.Chin.J.Catal.2002,23,9.[毛东森,卢冠忠,陈庆龄,谢在库,张玉贤.催化学报,2002,23,9.]
(39)Mao,D.S.;Lu,G.Z.;Chen,Q.L.;Xie,Z.K.;Zhang,Y.X.Catal.Lett.2001,77,119.doi:10.1023/A:1012787028360
(40)Wang,J.L.;Wang,K.C.;Cao,H.Y.;Chen,Y.D.;Liu,Z.M.;Zhu,Y.;Gong,M.C.;Chen,Y.Q.Acta P hys.-Chim.Sin.2009,25,689.[王健礼,王康才,曹红岩,陈永东,刘志敏,朱 艺,龚茂初,陈耀强.物理化学学报,2009,25,689.]doi:10.3866/PKU.WHXB200904211
