Be2+在水、甲醇和乙醇中结构性质的从头算分子动力学模拟
2013-10-18曾勇平杨正华
曾勇平 时 荣 杨正华
(扬州大学化学化工学院,江苏扬州 225002)
1 引言
金属离子在溶液中的溶剂结构及动力学性质方面的研究已经引起了广泛的关注.1-6由于离子的存在会强力扰动溶液的结构,因此在许多化学领域,如碱金属、碱土金属离子在水、甲醇等溶液中的性质已经采用实验和理论方法做了大量的研究.7-10这些研究工作主要集中在第一溶剂壳结构的稳定性、电荷转移以及光谱等方面.虽然离子的极化效应对溶液的影响会波及到第一溶剂壳很远的地方,但第一溶剂壳的性质仍是研究溶液性质的焦点.例如,在生物体内蛋白质通道对离子的选择性与第一溶剂壳的性质有很重要的关系.11近来,研究者们采用了准中子和弹性散射、12红外和拉曼光谱13-15以及飞秒光谱16来研究离子的溶剂化特征.这些实验方法研究的内容大多集中在宏观性质方面,对微观性质的了解还很有限,并存在着许多争议.而分子模拟方法已经被证明是一种描述分子微观性质的强有力工具,并用这些方法已经得到了一些微观性质方面的结果.经典的分子动力学(MD)和蒙特卡洛(MC)方法能够计算预测凝聚系统的平衡和非平衡性质,但此种方法没有传达任何电子结构方面的信息,另外缺乏普适性的经验力场,导致其相同体系不同力场对所研究结果的差异.而从头算分子动力学可以很好地克服这些问题,其仅有的假设是对离子运动作经典力学的近似,在最高水平的电子结构上完成系统基态电子性质的计算.由于从头算方法的时间和空间尺度都不够大,基于量子力学/分子力学(QM/MM)和可极化力场的MD方法也被广泛应用.17,18
离子半径对第一溶剂壳的配位数及稳定性有较大的影响,随着离子半径的增大,离子第一溶剂壳的稳定性降低,配位数增多.19Be2+是所有金属离子中半径最小的阳离子,在溶液中会紧紧束缚住溶剂壳周围的分子而表现出特殊的溶剂结构.水作为一种强的质子化溶剂有其独特的性质,甲醇是同时具有疏水基团(甲基)和亲水基团(羟基)的最小有机分子,其与水分子相比,结构上的差异使其具有两性特征.相对于甲醇分子,乙醇分子的疏水基团更大,Be2+在其中的溶剂结构又具有怎样的特征?而关于这方面研究尚未见报道.由此,水、甲醇和乙醇这三种分子在亲、疏水基团上的不同,可能表现出的结构差异引人关注.本文以水、甲醇和乙醇作为带有不同亲、疏水基团的分子对象,采用从头算分子动力学研究比较Be2+在不同的亲、疏水基团(甲基和乙基)极性溶剂中的微溶剂结构性质.
2 计算方法与体系模型
CPMD是基于第一性原理的分子动力学方法,它将密度泛函和分子动力学方法相结合,并最大限度地发挥了密度泛函的优势.本文采用Car-Parrinello分子动力学方法和局域密度近似赝势对Be2+在水、甲醇和乙醇的结构性质进行研究.为取得较大的时间步长,甲醇分子中的氢全部用同位数氘取代.金属离子选用Goedecker赝势,20C原子和O原子选用Martins-Troullier(MT)赝势,21计算赝势非局域部分的贡献采用Kleinman-Bylander方法.22H原子选用Von Barth-Car赝势.23这些赝势的选择在研究纯液体甲醇中得到了好的结果.24平面波截断能量选取为70 Ry(1 Ry=0.5 hartree),采用广义梯度近似(GGA)方法,用BLYP交换关联函数完成密度泛函计算.虚拟电子质量选取800 a.u.以使系统保持在玻恩-奥本海默面,运动方程积分时间步长为5 a.u.(0.12 fs).
体系模型根据分子在300 K温度下的密度确定(盒子尺寸及分子数见Supporting Information表S1)将一个Be2+置于研究体系中,计算采用周期性边界条件.首先采用全原子力场(OPLS)25,26进行分子动力学平衡1 ns,最后的平衡构型作为CPMD运行的初始构型.初始构型在300 K温度下以Car-Parrinello动力学热化3 ps,然后平衡2 ps,最后在NVE系综下模拟运行25 ps,取后12 ps作结果分析.经典分子动力学计算在实验室自己搭建的簇计算平台上完成,所有的CPMD计算在ScGrid的deepcomp7000上完成.对于纯流体,赝势选择以及能量截断都能够较好地与实验结果吻合,在以前的工作中已经报道.27为了验证Be2+的赝势的合理性,本文计算了Be2+分别与水、甲醇和乙醇二聚体的结构参数和结合能等方面的数据,经过比较与文献报道的部分实验及理论值较为一致.其二聚体的几何结构见图S1,二聚体分子间O…Be2+的距离、Be2+…OH角度及结合能数据汇总于表S2,从表中可以看出平面波基组的BLYP函数很好地重复了二聚体和络合物的结构参数.
3 结果与讨论
3.1 径向分布函数及第一溶剂壳的结构特征
径向分布函数(RDF)是研究结构性质的一种重要方法.其定义为:
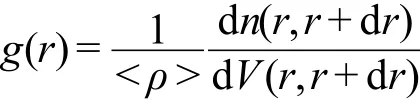
这里,d V=4πr2d r;d n是d V球壳内的原子数目;<ρ>为平均数密度,<ρ>=n/V.径向分布函数中第一个峰对应中心原子周围的第一个配位层(也即第一溶剂壳),第一个峰下面的面积(即g(r)从零积分到第一个最小值处)乘以<ρ>即为第一配位层中的原子数目N(即配位数).其表达式为:

图1 Be2+…O的径向分布函数(g(r))及相应的积分数(n(r))Fig.1 Radial distribution functions(g(r))and relative integration numbers(n(r))for Be2+…O
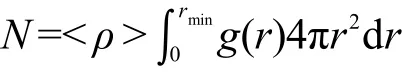
图1为水、甲醇和乙醇体系中Be2+…O径向分布函数及配位数.由图可以看出,径向分布函数第一个峰表现得都很显著,反映出分子对的排列呈近程有序性.且都出现明显第二峰,表明存在第二配位层.径向分布函数第一峰的最小值都为0,表明第一配位层与第二配位层之间分子交换较小,配位层较为稳定.Be2+…O径向分布函数第一个峰的位置落在同一个位置,为0.165 nm.由积分线可以得到第一溶剂壳的配位数为4,这说明了此第一溶剂壳为四配位的四面体结构.Yamaguchi等28采用X射线散色光谱得到BeCl2在水溶液中的配位距离为0.167 nm.Divjakovic等29采用实验得到Be(NO3)2溶液的配位距离为0.176 nm.Akitt等30采用NMR得到Be2+离子的配位数为4.1.Divjakovic等得到的配位距离略大外,其它实验结果与本文Be2+在水溶液中的第一溶剂壳的Be与O配位距离和配位数较为一致.Gnanakaran等31采用经典分子动力学得到径向分布函数的最高峰出现在(0.163±0.002)nm,配位数为4.Marx等32用从头算方法得到Be2+-水溶液第一溶剂壳中Be2+…O的距离为0.163 nm,配位数同样为4.Yang和Li等33采用经典分子动力学得到Be2+第一溶剂壳峰为0.167 nm,配位数为4.这些理论结果与本文得到Be2+在水中的第一溶剂壳峰和配位数的结果较为符合.对于Be2+在甲醇和乙醇中的径向分布函数及配位数尚无文献报道,在本文所研究的体系中,溶剂的变化对Be2+…O径向分布函数第一个峰的位置几乎没有影响,表明官能团差异没有引起显著的变化.这与Be2+本身特性密切相关.在液体水、甲醇、乙醇中,对溶液结构性质起决定作用的都为氢键,尽管溶剂发生了改变,但氧原子与Be2+作用力的本质不变.Be2+半径小,与周围溶剂氧原子作用强、束缚紧密,官能团影响处于次要的地位,因此不会影响第一个峰的位置及配位数.
三种体系的径向分布函数曲线第一个最小值在很大的范围内(0.193-0.313 nm)接近零,这表明以Be2+为球心,半径在0.193到0.313 nm的球壳内几乎没有氧原子出现,即这三体系中的第一溶剂壳结构都较为稳定.另一方面,由于Be2+的半径小,第一溶剂壳中的氧原子与Be2+结合得很强,溶剂分子进入或者离开第一溶剂壳较为困难.同时也造成第一溶剂壳中溶剂分子与体相中溶剂分子难以进行交换.径向分布函数曲线中峰高和峰宽是原子之间相互作用强度的标志.图中径向分布函数曲线,第一个峰的高度越高,宽度越窄,说明氧原子与Be2+结合越强,第一溶剂壳中溶剂分子的迁移率也越低.径向分布函数中第一个峰的峰宽几乎相同,但高度却有明显的差异,峰高由水到乙醇逐渐增高.这也说明第一溶剂壳稳定性顺序为:乙醇>甲醇>水.

图2 Be2+第一溶剂壳的瞬时构型Fig.2 Snapshots of the first solvation shell of Be2+

图3 第一溶剂壳中O―Be2+―O的角度分布Fig.3 O―Be2+―O angle distributions of the first solvation shell
为了能够清晰地说明第一溶剂壳中溶剂分子在Be2+周围的分布情况,图2给出了Be2+在三种体系中第一溶剂壳的瞬间构型.图2的结果与图1中的积分线所得到的配位数一致,这也直接证实了Be2+第一溶剂壳的配位数为4.从图中可以很明显地看出第一溶剂壳的构型为较为规整的四面体结构.为了验证第一溶剂壳四面体的准确性,本文统计了角度O―Be2+―O分布概率,图3为第一溶剂壳中O―Be2+―O的角度分布概率图.
第一溶剂壳中O―Be2+―O的角度分布只有一个很突出的主峰.对于每种溶剂,峰位置略有差异,但其峰的位置在109.5°附近,并且O―Be2+―O的角度变化范围较小,在95°到125°之间分布.Gnanakaran等31得到的O―Be―O的角度分布在87°到122°之间,分布极值范围略低于本文的结果,然而其得到最可能分布在109.5°,与本文一致.Marx等32得到∠O―Be―O最可能分布为110°,其角度分布范围在±25°范围内.这和本文得到Be2+在水中的角度分布一致,进一步证实Be2+第一溶剂壳为理想的四面体结构.
为了验证四面体结构是否稳定,图4为第一配位层中4个氧原子与Be2+之间的距离随着模拟时间演变的曲线.从图中可以看出,氧原子与Be2+之间的距离在0.165 nm左右波动,并且幅度很小,这与径向分布函数得到的结果一致(峰的位置为0.165 nm).在本文的模拟时间尺度内,第一溶剂壳中的溶剂分子都没有逃离第一溶剂壳,也没有与体相中的溶剂分子进行交换,表明Be2+在这三种溶液中的溶剂结构较为稳定.
图5为第一溶剂壳的距离范围内,配位数随着模拟时间的变化.由图可以看出,在本文所模拟的时间尺度内,这三种体系第一溶剂壳中的配位数都没有发生变化,同为4.表明在这段时间间隔内,没有溶剂分子逃离第一溶剂壳,没有其它的溶剂分子进来补充,这也与图4表现出的情形是符合的.导致这种情况发生的原因可能是Be2+的半径比较小,与溶剂壳氧原子形成了较强配位作用.
3.2 空间分布函数
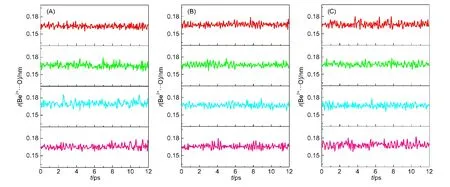
图4 Be2+与第一溶剂壳溶剂分子中O原子之间的距离随模拟时间的变化Fig.4 Be2+···O distances refer to solvent molecules in the first coordination shell along the simulation runs

图5 与Be2+距离在r(第一溶剂壳的范围)以内的氧原子数目随着模拟时间的变化(即配位数)Fig.5 Number of oxygen atoms(i.e.,coordination number in the first solvation shell)within a radius of r from the Be2+along the simulation run

图6 第一溶剂壳中Be2+在溶剂分子周围的空间分布函数Fig.6 Spatial distribution of Be2+relative to solvent molecules in the first solvation shell

图7 第一溶剂壳中氧原子在Be2+周围的空间分布函数Fig.7 Spatial distribution of oxygen atoms relative to Be2+ion in the first solvation shell
为了给出溶液结构详细的局部信息,以及同等距离上的粒子在不同位置分布密度的高低,本文计算了原子的空间分布函数.图6给出了第一溶剂壳中Be2+在溶剂分子周围的空间分布函数图.可以清晰地看出,Be2+在溶剂分子的等高面上并不是均匀的分布,主要集中分布在溶剂分子接受氢键的方向.在相对强度比较高时,Be2+在水分子和乙醇分子周围主要分布在两个区域,而在甲醇分子周围则主要集中在一个区域.由于水分子中的一个氧原子可以接受两个氢原子,形成两个氢键,因此Be2+在氧原子接受形成氢键的两个地方出现的概率较大.而对于甲醇,一个分子只能接受一个氢原子形成氢键,因此Be2+的分布主要集中在一个区域.对于乙醇分子,虽然一个乙醇分子只能接受一个氢原子形成氢键,但乙醇分子有两种形式的稳定构象(邻位交叉构象),即羟基的氢原子有两种指向可以形成邻位交叉构象的稳定形式,所以Be2+会在乙醇分子周围两个区域有较高分布概率,因此呈现出与甲醇不一样的结构定位.
图7为第一溶剂壳中氧原子在Be2+周围的空间分布函数.图中氧原子主要集中分布在四面体的四个顶端附近,在其它区域分布的概率较低.根据氧原子在Be2+周围的分布,可以明显地看出配位数都为4,所以氧原子也主要集中分布在Be2+周围的四个区域,进一步证实了溶剂壳的稳定性.对于这三种溶剂,Be2+-水溶液中,氧原子在Be2+周围分布相对集中一些,定域性更强.虽然同为4个区域,但在Be2+-甲醇溶液和Be2+-乙醇溶液中,氧原子的分布则稍广.与水相比,Be2+在甲醇和乙醇溶剂壳中空间分布函数分布较广,随着官能团的变化,这种差异更加明显.表明官能团的差异对局域第一溶剂壳造成了一定的影响,官能团较大,其配位结构也较为柔性.
4 结论
采用CPMD方法计算了Be2+在水、甲醇和乙醇的径向分布函数、配位数和空间分布函数等结构性质.考察了基团极性对流体结构的影响.计算结果表明在径向分布函数中,溶剂的差异对Be2+…O第一个峰的位置影响很小.三个体系的径向分布函数曲线第一个最小值在0.193-0.313 nm范围内,接近零,表明Be2+在这三个体系中的第一溶剂壳结构都非常稳定.另一方面,由于Be2+的半径很小,第一溶剂壳中的氧原子与Be2+作用较强,体相分子与第一溶剂壳分子之间的交换较为困难.对溶剂壳层结构分析,统计O―Be2+―O分布概率发现,第一溶剂壳中O―Be2+―O的角度分布只有一个很突出的主峰.每种溶剂峰位置有较小差异,这些峰的位置在109.5°附近,并且O―Be2+―O的角度变化范围较小,证实了Be2+第一溶剂壳为理想的四面体结构.通过分析空间分布函数表明Be2+在溶剂分子的等高面上并不是均匀地分布,金属离子主要集中分布在溶剂分子接受氢键的方向.在相对强度比较高时,Be2+在水分子和乙醇分子周围主要分布在两个区域,而在甲醇分子周围则主要集中在一个区域.
Supporting Information∶The simulation system size and the calculated structure properties,binding energies of optimized geometries of dimer have been included.This information is available free of charge via the internet at http://www.whxb.pku.edu.cn.
(1)Rempe,S.B.;Pratt,L.R.;Hummer,G.;Kress,J.D.;Martin,R.L.;Redondo,A.J.Am.Chem.Soc.2000,122,966.
(2)Liu,Y.;Wang,F.F.;Yu,C.Y.;Liu,C.;Gong,L.D.;Yang,Z.Z.Acta Phys.-Chim.Sin.2011,27,379.[刘 燕,王芳芳,于春阳,刘 翠,宫利东,杨忠志.物理化学学报,2011,27,379.]doi:10.3866/PKU.WHXB20110233
(3)Rudolph,W.W.;Fischer,D.;Irmer,G.;Pye,C.C.Dalton Transactions 2009,33,6513.
(4)Lyubartsev,A.P.;Laasonen,K.;Laaksonen,A.J.Chem.P hys.2001,114,3120.doi:10.1063/1.1342815
(5)Ayala,R.;Martinez,J.M.;Pappalardo,R.R.;Muňoz-Paez,A.;Marcos,E.S.J.P hys.Chem.B 2008,112,5416.doi:10.1021/jp076032r
(6)Jiao,D.;King,C.;Grossfield,A.;Darden,T.A.;Ren,P.J.P hys.Ch em.B 2006,110,18553.doi:10.1021/jp062230r
(7)Spangberg,D.;Hermansson,K.Chem.Phys.2004,300,165.
(8)Ansell,S.;Barnes,A.C.;Mason,P.E.;Neilson,G.W.;Ramos,B.S.Biophysical Ch emistry 2006,124,171.doi:10.1016/j.bpc.2006.04.018
(9)Megyes,T.;Radnai,T.;Grósz,T.;Pálinkás,G.Journal of Molecular Liquids 2002,101,3.doi:10.1016/S0167-7322(02)00098-3
(10)Faralli,C.;Pagliai,M.;Cardini,G.;Schettino,V.J.P hys.Chem.B 2006,110,14923.
(12)Herdman,G.J.;Salmon,P.S.J.A m.Chem.Soc.1991,113,2930.doi:10.1021/ja00008a022
(13)Guillot,B.;Marteau,P.;Obriot,J.J.Chem.Phys.1990,93,6148.doi:10.1063/1.458986
(14)Cory,C.;Pye,W.W.;Rudolph,J.Phys.Chem.1998,102,9933.doi:10.1021/jp982709m
(15)Yu,X.C.;Lin,K.;Hu,N.Y.;Zhou,X.G.;Liu,S.L.Acta P hys.-Chim.Sin.2010,26,2473.[余小春,林 珂,胡乃银,周晓国,刘世林.物理化学学报,2010,26,2473.]doi:10.3866/PKU.WHXB20100922
(16)Omta,A.W.;Kropman,M.F.;Woutersen,S.Bakker,H.J.Science 2003,301,347.doi:10.1126/science.1084801
(17)Rode,B.;Hofer,T.;Randolf,B.;Schwenk,C.;Xenides,D.;Vchirawongkwin,V.Theor.Chem.A cc.2006,115,77.doi:10.1007/s00214-005-0049-1
(18)Yu,H.B.;Whitfield,T.W.;Harder,E.;Lamoureux,G.;Vorobyov,I.;Anisimov,V.M.;MacKerell,A.D.;Roux,B.J.Chem.Theory Comput.2010,6,774.doi:10.1021/ct900576a(19)Chowdhuri,S.;Chandra,A.J.Chem.P hys.2006,124,084507.doi:10.1063/1.2172598
(20)Goedecker,S.;Teter,M.;Hutter,J.Phys.Rev.B 1996,54,1703.doi:10.1103/PhysRevB.54.1703
(21)Troullier,N.;Martins,J.L.P hys.Rev.B 1991,43,1993.doi:10.1103/PhysRevB.43.1993
(22)Kleinman,L.;Bylander,D.M.Phys.R ev.L ett.1982,48,1425.doi:10.1103/PhysRevLett.48.1425
(23)Vuilleumier,R.;Sprik,M.J.Ch em.Phys.2001,115,3454.doi:10.1063/1.1388901
(24)Pagliai,M.;Cardini,G.;Righini,R.;Schettino,V.J.Chem.Phys.2003,119,6655.doi:10.1063/1.1605093
(25)Jorgensen,W.L.;Maxwell,D.S.;Tirado-Rives,J.J.Am.Chem.Soc.1996,118,11225.doi:10.1021/ja9621760
(26)Kaminski,G.;Friesner,R.A.;Tirado-Rives,J.;Jorgensen,W.L.J.Phys.Chem.B 2001,105,6474.doi:10.1021/jp003919d
(27)Zeng,Y.P.;Zhu,X.M.;Yang,Z.H.Acta Phys.-Chim.Sin.2011,27,2779.[曾勇平,朱小敏,杨正华.物理化学学报,2011,27,2779.]doi:10.3866/PKU.WHXB20112779
(28)Yamaguchi,T.;Ohtaki,H.;Spohr,E.;Palinkas,G.;Heinzinger,K.;Probst,M.M.Zeitschrift Fur Naturforschung Section A-AJournal of Ph ysical Sciences 1986,41,1175.
(29)Divjakovic,V.;Endenharter,A.;Nowacki,W.;Ribar,B.Z.Kristallogr.1976,144,314.doi:10.1524/zkri.1976.144.1-6.314
(30)Akitt,J.W.;Duncan,R.H.J.Chem.S oc.Farad ay Trans.1980,76,2212.doi:10.1039/f19807602212
(31)Gnanakaran,S.;Scott,B.;McCleskey,T.M.;Garcia,A.E.J.Phys.Chem.B 2008,112,2958.doi:10.1021/jp076001w
(32)Marx,D.;Sprik,M.;Parrinello,M.Chem.Phys.Lett.1997,273,360.doi:10.1016/S0009-2614(97)00618-0
(33)Yang,Z.;Li,X.J.Chem.Phys.2005,123,094507.doi:10.1063/1.2000245
