Cuギ-N,N-二(2-羧基苯基)-2,6-吡啶二甲酰胺配合物的光谱性质及结构
2013-10-17王彩荣王璟琳杨斌盛
王彩荣 王璟琳 刘 斌 杨斌盛*,
(1山西大学分子科学研究所,化学生物学与分子工程教育部重点实验室,太原 030006)
(2长治学院化学系,长治 046011)
CopC是由102个氨基酸残基构成的可溶性膜间蛋白,是假单胞菌体内铜调控蛋白之一[1]。NMR和EXAFS研究表明[2-3],CopC蛋白在溶液中呈现由9股β折叠围成的桶状结构,2个相距约为3 nm的铜ガ、铜ギ结合部位位于桶状结构的两端。生理pH条件下的Cuギ-CopC-Cuガ晶体结构[4]表明,位于C端的铜ガ与48位组氨酸残基、40位甲硫氨酸残基配位,位于N端的铜ギ与1、91位组氨酸残基咪唑氮、N端氨基氮、89位天冬氨酸残基的羧基氧及水分子中的氧原子配位,形成五配位的畸变四方锥结构。CopC在生物体内铜离子的转运过程中起氧化还原开关作用[5-9],然而其作用机理尚不清楚。
2006年Gudasi等[10]首次报道了一种含有羧基的吡啶酰胺类配体 N,N-二(2-羧基苯基)-2,6-吡啶二甲酰胺(BCPD)与Cuギ离子在醇水混合体系中的配位作用,推测BCPD与2个Cuギ同时作用,形成了双核1∶2型配合物。本文试图选取BCPD作为CopC蛋白的模型化合物,研究生物体系中Cuギ和Cuガ离子配位模式和氧化还原过程。有别于该文献的是,通过元素分析、吸收光谱、荧光光谱和X-射线晶体衍射等方法发现,该配体只结合一个Cuギ离子,形成了1∶1型五配位的畸变四方锥结构。BCPD与金属离子的配位模式呈现出了多样性。
1 实验部分
1.1 试剂和仪器
2,6-吡啶二甲酸(PDA)(北京百灵威化学技术有限公司),邻氨基苯甲酸(湖南湘中地质实验研究所,使用前重结晶),水合醋酸铜(Cu2(CH3COO)4·2H2O,简写为 CuAc2·H2O),氯化铜(CuCl2·2H2O)以及其它试剂均为分析纯。实验用水为二次蒸馏水。
Perkin-Elmer 240C型元素分析仪;Bruker SMART 1000 CCD X-衍射仪 (德国Bruker公司;HP 8453型UV-Vis吸收光谱仪;Hitachi-850型荧光光谱仪。
1.2 配体BCPD的合成
参照文献[10]:将 2,6-吡啶二甲酸(1.67 g,10 mmol)加入25 mL新蒸馏的二氯亚砜中,加热回流5~6 h直至溶液变得澄清。冷却到室温,减压蒸馏除去过量的二氯亚砜,得到产物吡啶-2,6-二甲酰氯,产物不经分离直接用于后续反应。将重结晶过的2-氨基苯甲酸(2.74 g,20 mmol)溶于干燥的四氢呋喃中,在冰水浴中不断搅拌下,慢慢滴入上述制备的吡啶-2,6-二甲酰氯中。室温下反应2 h,冷却,过滤,沉淀用甲醇洗涤,DMF重结晶,得到浅黄色固体2.9 g产品,产率71%。
产品为淡黄色固体粉末。M.P.值为 278~280℃。元素分析实测值 (%,C21H15N3O6计算值):N 10.08(10.37),H 4.004(3.73),C 61.98(62.22)。1H NMR(DMSO-d6)中 δ值:13.39(2H,s,COOH),12.94(2H,s,NH),8.70(2H,d,吡啶 β-H),8.25(H,s,吡啶 γ-H),8.0(2H,d,吡啶 α-H),7.25~8.33(8H,m,苯环 H)。
1.3 Cu2+离子对BCPD的光谱滴定
室温条件下,配制BCPD的乙醇溶液时滴加少量三乙胺使其完全溶解(pH=7~8),以 CuAc2·H2O(1.0 mmol·L-1)的乙醇溶液对 BCPD(40 μmol·L-1)的乙醇溶液进行滴定,分别利用HP 8453型UV-Vis吸收光谱仪和Hitachi-850型荧光光谱仪测定光谱。
1.4 铜配合物的合成
称取 81 mg(0.2 mmol)BCPD,加入 15 mL 的甲醇中,滴加少量三乙胺助溶(pH=7~8),再加入40 mg(0.2 mmol)CuAc2·H2O,溶液呈现深绿色。 搅拌下加热回流2 h,采用自然蒸发溶剂法,得到深绿色单晶。元素分析实测值 (%,C34H47N5O7Cu,计算值):N 10.08(9.99),H 6.73(6.75),C 58.37(58.23)。
1.5 配合物晶体结构测定
选取大小为 0.47 nm×0.44 nm×0.35 nm 的单晶于Bruker SMART 1000 CCD单晶X射线衍射仪上,在298(2)K下使用经石墨单色化的Mo Kα射线(λ=0.071 073 nm) 为辐射光源, 在 2.60°<θ<25.02°范围内,以φ-ω扫描方式采用SMART软件[11]收集衍射点17 497个。选取I>2σ(I)的6404个可观察点用于结构测定和修正[12],衍射数据均经LP因子和经验吸收校正。先用直接法求出结构,后用全矩阵最小二乘法F2进行修正,找出全部非氢原子的坐标,做各向异性温度因子处理,直至收敛。所有氢原子都采用理论加氢法。π…π相互作用距离和位移角使用DIAMOND[13]程序计算,相关图形使用SHELXTL-97程序[14]和DIAMOND程序进行绘制。晶体学数据列于表1。
CCDC:911182。

表1 配合物BCPD-Cu(II)的晶体学数据Table 1 Crystallographic data and structure refinement for complex(BCPD-Cu)

续表1
2 结果与讨论
2.1 光谱性质
2.1.1 紫外差光谱
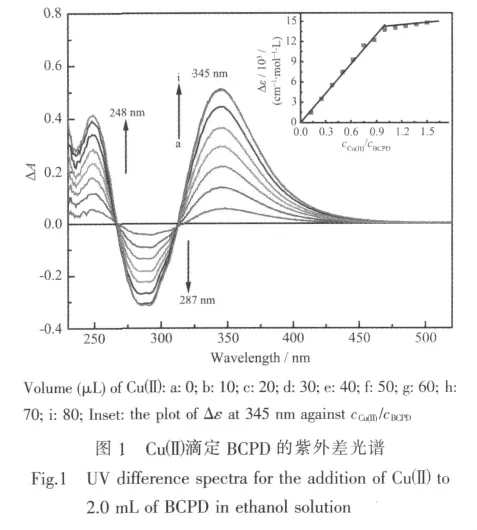
图 1 为乙醇体系中 CuAc2·H2O(1.0 mmol·L-1)滴定 BCPD(40 μmol·L-1)溶液的紫外差光谱。由图 1可见,随着Cuギ的加入,差光谱在248、345 nm两处出现最大正吸收峰,在287 nm处出现1个负吸收峰,在266和312 nm处有2个等吸收点。当Cuギ滴加到一定量时,光谱不再发生明显变化。将λ=345 nm处对应的吸光度除以BCPD的分析浓度,得出BCPD与Cuギ结合的表观摩尔吸光度(Δε)。将Δε对cCuギ/cBCPD作图如图1插图。从图1可见,Δε随cCuギ/cBCPD的增加而线性增加,在cCuギ/cBCPD=1.0处出现明显拐点。表明Cuギ与BCPD之间形成1∶1型配合物(简写为Cuギ-BCPD),该滴定曲线的斜率即为λ=345 nm处Cuギ-BCPD配合物的摩尔吸光系数,经线 性 拟 合 可 得 到 ΔεBCPD-Cu=(14.59±0.35)×103cm-1·mol-1·L。
2.1.2 荧光光谱
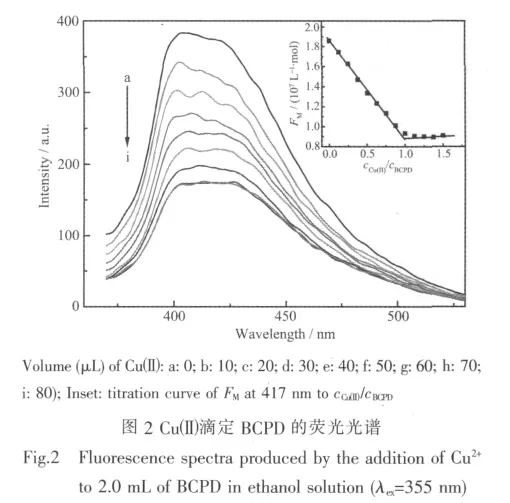
图 2 为 CuAc2·H2O(1.0 mmol·L-1)滴定 BCPD(40 μmol·L-1)溶液的荧光光谱。配体BCPD在405~420 nm之间出现一宽的荧光峰,随着Cuギ的滴加,BCPD的荧光逐渐被猝灭。当Cuギ滴加到一定量时,荧光强度不再发生明显变化。为了消除稀释效应,将417 nm处对应的荧光强度除以BCPD的分析浓度,得出表观摩尔荧光强度FM。将FM对cCuギ/cBCPD作图,如图2插图。FM随cCuギ/cBCPD的增加而线性减小, 在 cCuギ/cBCPD=1.0 处出现拐点, 在 cCuギ/cBCPD>1.0后,FM不再随cCuギ/cBCPD的增加而变化,表明 Cuギ与BCPD之间形成稳定的配合物,其结合比为1∶1。这与紫外差光谱得出的结果一致。
2.1.3 结合常数
在乙醇溶液中配体BCPD与Cuギ的总浓度恒定为 20 μmol·L-1的条件下,改变溶液中 cBCPD与 cCuギ的比例并以相同浓度的BCPD溶液作参比,测定溶液在 345 nm 处的吸光度,将 A345nm对 XCuギ[XCuギ=cCuギ/(cCuギ+cBCPD)]作图,结果见图 3。从图 3 可知,A345nm随着XCuギ的增加先增大后又减小,整个曲线经拟合呈抛物线,将两侧切线反向延长交于一点,该点对应的XCuギ在 0.5 附近,表明该体系中 BCPD 与 Cuギ以 1∶1的形式配位, 配合物的稳定常数为 K=(4.59±0.05)×106mol-1·L。

2.2 可见吸收光谱分析
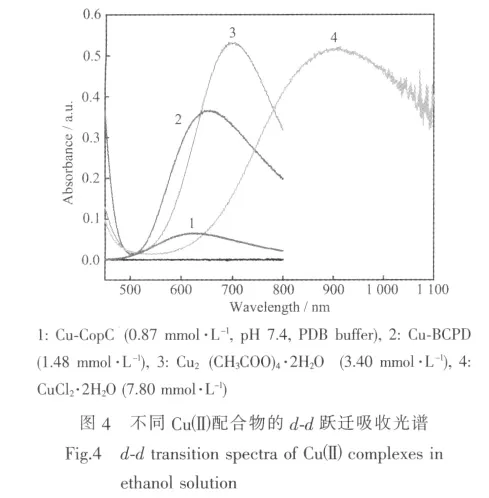
Cuギ的配体场不同,d轨道的能级分裂程度就不同。图4所示为不同Cuギ配合物在乙醇溶液及Cuギ-CopC 在 pH 7.4,20 mmol·L-1磷酸缓冲溶液中的d-d跃迁吸收光谱。从图4可见:配合物Cuギ-CopC,Cu ギ-BCPD,Cu2(CH3COO)4·2H2O 和 CuCl2·2H2O 对应 d-d 跃迁分别位于 615 nm(ε=76 mol-1·L·cm-1),645 nm(ε=246 mol-1·L·cm-1),699 nm(ε=157 mol-1·L·cm-1)和 901 nm(ε=66 mol-1·L·cm-1)处。 可见这4类Cuギ配合物的配体场由强至弱的顺序为Cuギ-CopC>Cuギ-BCPD>Cu2(CH3COO)4·2H2O>CuCl2·2H2O;随着配体场效应减弱,对应的d-d跃迁吸收峰逐渐红移。
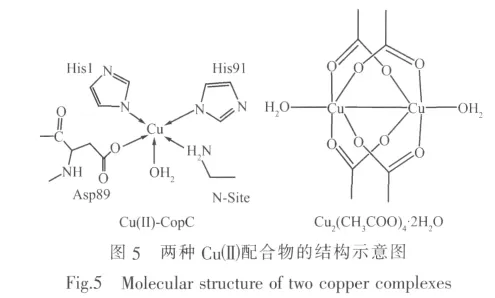
在 Cuギ-CopC中 (图 5),Cuギ分别与 2个组氨酸残基咪唑氮、N端氨基氮、水分子中的氧及天冬氨酸残基的羧基氧配位,形成五配位的畸变四方锥构型,天冬氨酸残基的羧基O原子占据四方锥的顶点,属于Cuギ-N3O2的配位结构。而Cu2(CH3COO)4·2H2O中(见示意图5),Cuギ与羧基氧和水分子配位,属于Cuギ-O5配位结构。Cuギ)-BCPD中Cuギ的d-d跃迁吸收光谱与Cuギ-CopC的d-d跃迁谱比较接近,说明二者配位环境相似。故推测本文中Cuギ-BCPD中Cuギ也倾向于和BCPD中吡啶氮,酰胺氮和羧基氧配位。Cu-BCPD配合物的晶体结构分析验证了这一推测。
2.3 铜配合物结构分析
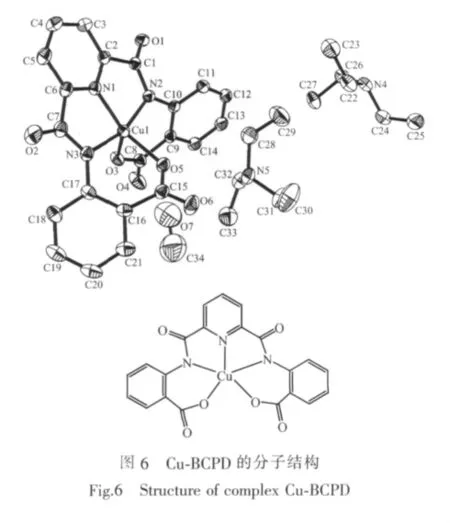
通过三乙胺调节溶液的pH,在甲醇中利用CuAc2·2H2O和BCPD反应,得到了Cu-BCPD配合物晶体。目标配合物的分子结构如图6所示。主要键长和键角数据见表2。晶体结构表明,标题配合物的分子式为:[Cu(C21H11O6N3)]2-·2Et3NH+·CH3OH,中心离子Cuギ与配体BCPD中的3个N原子(N(1),N(2),N(3))和2个O原子(O(3),O(5))形成五配位畸变的四方锥构型,顶点位置被O(3)原子所占据。我们发现,配体中酰胺基团的2个氮原子N(2)和N(3)采取了脱质子形式与Cuギ配位:首先,N(2)原子与相邻C1、C10和Cu1间的夹角均接近120℃,分别为C(10)-N(2)-Cu(1)=123.2(3),C(1)-N(2)-C(10)=119.0(4),C(1)-N(2)-Cu(1)=116.7(3),N(3)与 相 邻 的 C(7)、C(17)和Cu(1)间夹角总和也是359.9℃,说明此时的酰胺N原子是sp2杂化模式形成平面三角形结构;同时,二面角 N(1)-Cu(1)-N(2)-C(1)=-3.2(3),N(1)-Cu(1)-N(3)-C(7)=-1.2(3)°近于 0 ℃,说明 Cu(I)、N(2)、N(3)、C(1)和C(7)均与吡啶环几乎共平面;N(1)-Cu(1)-N(2)-C(10)=-171.7(4)°,N(1)-Cu(1)-N(3)-C(17)=-177.5(4)°,说明两个苯环上的C(10)和C(17)也与吡啶环几乎共平面;另外,配合物中 N(2)-C(1),N(3)-C(7)的键长小于对应 N(2)-C(10)和 N(3)-C(17)的 键 长,C(1)-O(1),C(7)-O(2)的键长大于典型羰基 C=O(0.122 nm)的键长,这表明N(2)、N(3)脱去质子后与羰基形成局部离域体系[N(2)C(1)O(1)]-和[N(3)C(7)O(2)]-[17]。由此可以推论酰胺基团中的2个氮原子N(2)和N(3)采用脱质子形式与Cuギ配位。配离子带2个单位负电荷,外界为2个质子化的三乙胺和1个溶剂分子CH3OH。由于这种特殊的配位方式,Cu-BCPD可以被作为Cu-CopC的一个分子模型来研究。
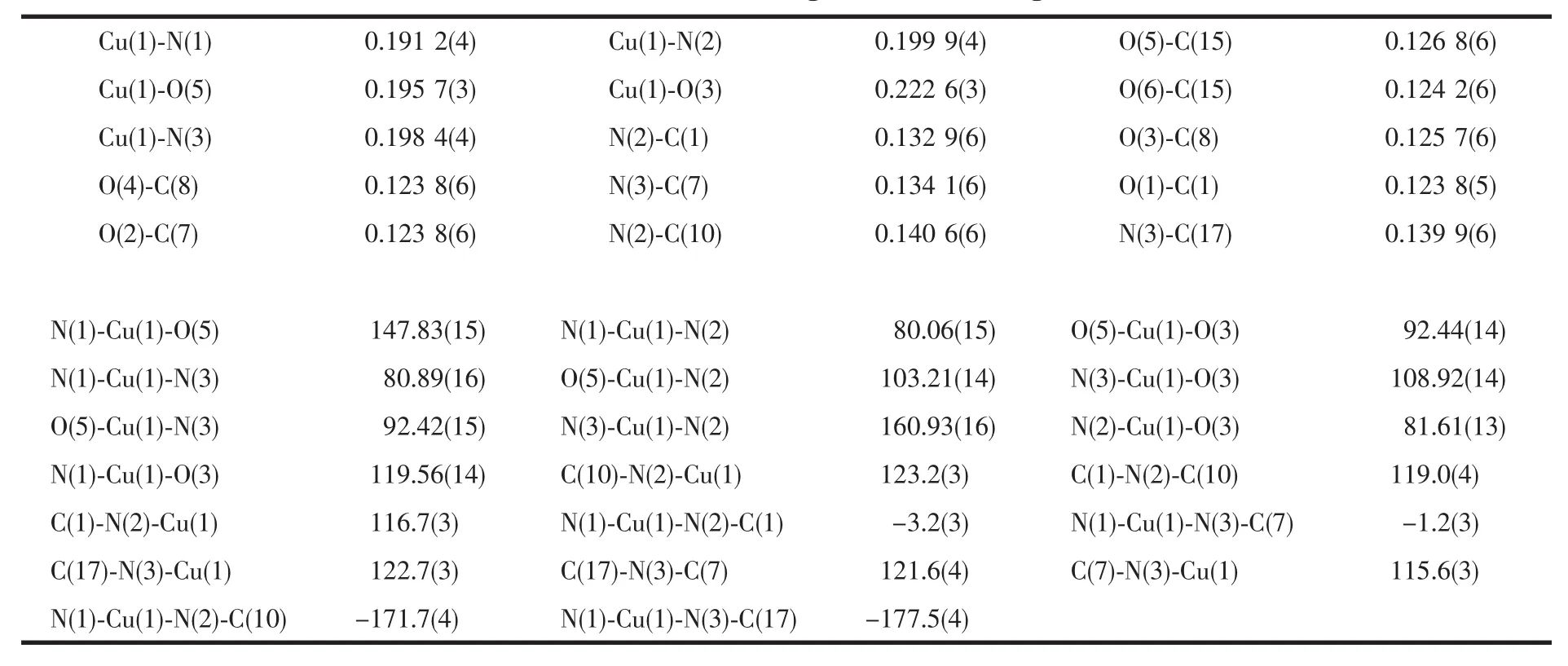
表2 配合物的主要键长和键角Table 2 Selected bond lengths(nm)and angles(°)
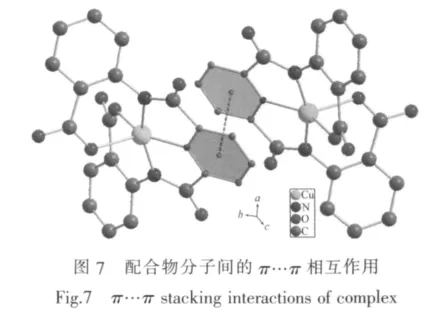
吡啶环(N(1)、C(2)~C(6))与苯环(C(9)~C(14))、(C(16)~C(21))之间的二面角分别为 44.6°和 33.6°,2 个苯环之间的二面角为57.1°。目标配合物分子间存在π-π相互作用见图7,为清晰起见,略去氢原子、外界阳离子Et3NH+和溶剂分子。2个分子中的吡啶环平面接近平行。面心距离为0.377 56(2)nm,两平面之间的垂直距离(即法线)为0.356 92 nm,面心距离与法线之间的夹角θ为19.0°。
与本文不同的是,文献[10]合成的BCPD-M2金属配合物 (M=Cu2+,Ni2+,CO2+,Mn2+,Zn2+,Cd2+)中 BCPD与2个Cuギ同时作用,形成了双核1∶2型配合物。这是由于反应条件的不同所致。文献所述的化合物是在VEtOH/VH2O=1∶1的混合体系中合成 (pH=9~10)的,而本文所述均是在醇体系中完成(pH=7~8)。可能是由于在醇等弱极性体系下,配体BCPD的酰胺氮原子容易脱去质子参与配位,而羰基氧原子不易配位。可见溶剂的极性和酸碱度对BCPD配位模式会产生不同影响。
3 结 论
本文通过紫外-可见吸收光谱、荧光光谱、单晶X射线衍射等方法研究了Cuギ离子与BCPD在醇溶液中的结合作用。实验表明,在醇溶液中(pH=7~8)二者形成了1∶1型配合物,表观结合常数为(4.59±0.05)×106mol-1·L; 晶体结构表明,Cuギ与 BCPD 分子的吡啶氮原子、羧基氧和去质子化的酰胺氮原子配位,形成五配位畸变四方锥结构。可见溶剂的极性或酸碱度影响了BCPD酰胺氮原子和羰基氧原子的配位模式,与金属离子的配位模式呈现出了多样性。
[1]Silver S.Gene,1996,179:9-19
[2]Arnesano F,Banci L,Bertini I,et al.PNAS,2003,100(7):3814-3819
[3]Arnesano F,Banci L,Bertini I,et al.Structure,2002,10:1337-1347
[4]Zhang L,Koay M,Maher M J,et al.J.Am.Chem.Soc.,2006,128(17):5834-5850
[5]Puig S,Rees E,Thiele D J.Structure,2002,10:1292-1295
[6]Li H Q,Zhao Y Q,Zheng X Y,et al.Spectrochim.Acta Part A,2009,72(1):56-60
[7]Li H Q,Zhao Y Q,Yang B S.Chin.J.Chem.,2009,27:1762-1766
[8]Zheng X Y,Pang E G,Yang B S,et al.Supramol.Chem.,2008,20(6):553-557
[9]Zheng X Y,Pang E G,Li H Q,et al.Chin.Sci.Bull.,2007,52(6):743-747
[10]Gudasi K B,Patil S A,Vadavi R S,et al.Polish.Chem.J.,2006,80:247-257
[11]SMART(Version 5.0)and SAINT(Version 6.02);Bruker AXS Inc.:Madison,Wisconsin,USA,2000.
[12]SheldrickG M.SADABS,A Program forAbsorption Corrections,University of Gottingen,Germany,1996.
[13]Klaus B.DIAMOND,Version 1.2c,University of Bonn,Germany,1999.
[14]Sheldrick G M.SHELXS 97 and SHELXTL 97,University of Gottingen,Germany,1997.
[15]LUO Shi-Xia(罗世霞),CHEN Xiao-Liang(陈晓靓),ZHU Huai-Wu(朱 淮 武 ),et al.J.Instrum.Anal.(Fenxi Ceshi Xuebao),2012,31(2):211-215
[16]Jain S L,Bhattacharyya P,Milton H L,et al.J.Chem.Soc.,Dalton Trans.,2004:862-871
[17]YANG Zheng-Yin(杨正银),YANG Ru-Dong(杨汝栋),YU Kai-Bei(郁 开 北 ).Acta Chim.Sinica(Huaxue Xuebao),1999,57(3):236-243
