分子模拟技术在脂肪酶性质及催化机理研究中的应用进展
2013-10-11王哲,王普,黄金
王 哲,王 普,黄 金
(浙江工业大学生物制药研究所,浙江 杭州 310014)
脂肪酶(lipase,EC 3.1.1.3)是一种甘油酯水解酶,属α/β折叠水解酶类。它广泛存在于动物、植物和微生物细胞中。脂肪酶不仅可在油水两相界面催化天然底物甘油酯的水解,还可在疏水介质中催化酯化、转酯化、酯交换等反应。由于脂肪酶具有反应活性高、立体选择性强和稳定性好等优点,使其在食品、皮革、造纸、有机合成、油脂化工、洗涤剂、化妆品、医药以及生物柴油合成等诸多领域具有广泛应用。近年来,有关脂肪酶催化机理研究已逐渐引起人们的关注。脂肪酶有别于酯酶、蛋白酶等其它α/β折叠水解酶的最显著特征在于它的催化活性中心部位被一个或多个α-螺旋片段遮盖(称为“盖子”结构)。这种特殊结构使得当环境中存在油水两相界面时,盖子结构打开,从而暴露出脂肪酶催化活性中心,发挥其催化功能(在行使催化功能时,脂肪酶的催化活性中心三元组中的组氨酸(His)从丝氨酸(Ser)上吸收一个质子,使得Ser亲核攻击羰基上的碳原子,形成特殊的脂肪酶-底物复合物的四面体过渡态 TS(tetrahedral transition state)(见图1)。在整个过程中天冬氨酸(Asp)通过与组氨酸(His)形成氢键起到稳定组氨酸位置的作用)。

随着计算机技术、生物信息学和结构生物学等研究领域的快速发展,利用分子模拟技术研究脂肪酶的结构特点,在催化过程中的动态结构变化进而理解其反应机理和构效关系,最终实现对脂肪酶的理性设计与改造已引起众多学者浓厚的研究兴趣[1-2]。分子模拟技术的发展进程大致可分为3个阶段:早期的皮秒(ps)级别的分子动力学模拟;柔性的分子对接技术;运算时间更长的分子动力学模拟以及 QM/MM(quantum mechanics/molecular mechanics,量子力学/分子力学)。
1 早期的分子动力学模拟
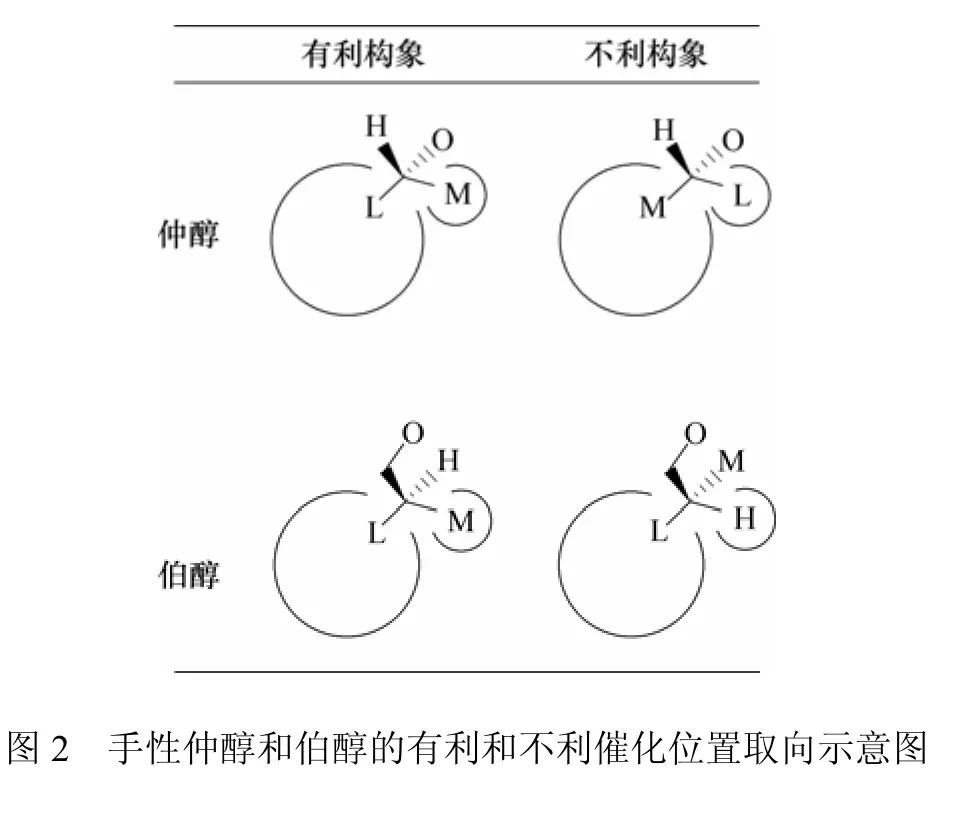
分子动力学模拟技术[3]主要依靠牛顿力学来模拟分子体系的运动,从而获得生物大分子在已知最低能量构象附近一系列合理的空间构象。因此,早期关于脂肪酶分子动力学模拟技术主要是用于脂肪酶构象变化的研究。Norin等[4]于1994年通过分子动力学方法比较了米黑根毛霉脂肪酶(Rhizomucor mieheilipase,RmL)在真空、水溶液和非极性溶液等不同溶剂条件下的构象变化,发现RmL 在非极性有机溶剂中比水相中的刚性更强,并且依据在乙酸乙酯溶剂中的动力学模拟,揭示出RmL 盖子结构的关闭与打开状态。Peters等[5]1996年研究多种溶剂对RmL结构的影响时也得出类似的结论。Zuegg等[6]于1997年运用分子动力学模拟方法研究了褶皱假丝酵母脂肪酶(Candida rugosalipase,CrL)和洋葱假单胞菌脂肪酶(Pseudomonas cepacialipase,PcL)两种脂肪酶催化水解伯醇酯和仲醇酯的四面体过渡态构象差异,提出了“大小口袋”规则(见图2),并依据该规则揭示了脂肪酶催化上述两种酯水解的对映体选择性机理。由于受到当时计算机的运算速度所限,整个分子动力学模拟过程仅运行了15 ps。
2 柔性分子对接技术预测脂肪酶的对映体选择性及催化机理
分子对接技术是通过几何匹配、能量匹配和化学环境匹配来寻找复合物的最优状态[7]。各种分子对接方法的计算体系均存在不同程度的简化。根据简化的程度和方式不同,可将分子对接方法分为刚性对接、半柔性对接和全柔性对接,其中,刚性对接主要用于蛋白-蛋白对接。随着分子对接技术的发展,人们开始运用半柔性或全柔性的对接技术研究脂肪酶对底物的对映体选择性。Tafi等[8]将全柔性对接技术应用于定性预测脂肪酶水解反应的对映体选择性研究,通过分子对接打分以及复合物的几何构象是否符合脂肪酶过渡态构象来判断立体选择性的优劣(见图3)。
柔性分子对接技术具有快速预测脂肪酶在催化过程中的立体选择性的优点,因此被广泛应用于脂肪酶对底物选择性的高通量虚拟筛选[9-13]。Tyagi等[9]通过对分子对接打分,预测了6种脂肪酶水解12种底物的能力(见表1),但由于仅依据对接打分来进行预测,其评价体系过于单一且导致预测结果并不理想,需要进一步结合复合物几何构象等因素综合判断对接结果。Xu等[10]通过设计南极假丝酵母脂肪酶(Candida antarcticalipase B,CaLB)催化底物四面体过渡态的几何构象来定性预测该脂肪酶对底物的对映体选择性。经验证,233个化合物中有 223个化合物被正确识别,预测准确率高达95.7%。分子对接技术作为一种简单快速的计算机模拟手段应用于定性预测脂肪酶催化立体选择性的研究已有较多报道,但其计算精确度不高,不能全面地阐述脂肪酶的催化机理。因此分子对接方法多被用于模拟初始的复合物构象,再结合运用其它手段,如定量构效关系(QSAR)、分子动力学和QM/MM 等方法,进一步研究其催化机理。
3 长运算时间的分子动力学模拟、QM/MM 方法及QSAR方法
随着计算机运算能力的发展,分子动力学方法由最初只能计算皮秒(ps)级别的分子运动,逐渐发展到纳秒(ns)级、甚至是微秒(μs)级。在此期间,人们利用分子动力学方法研究脂肪酶性质及催化机理的报道逐渐增多,且预测结果也越来越精准。
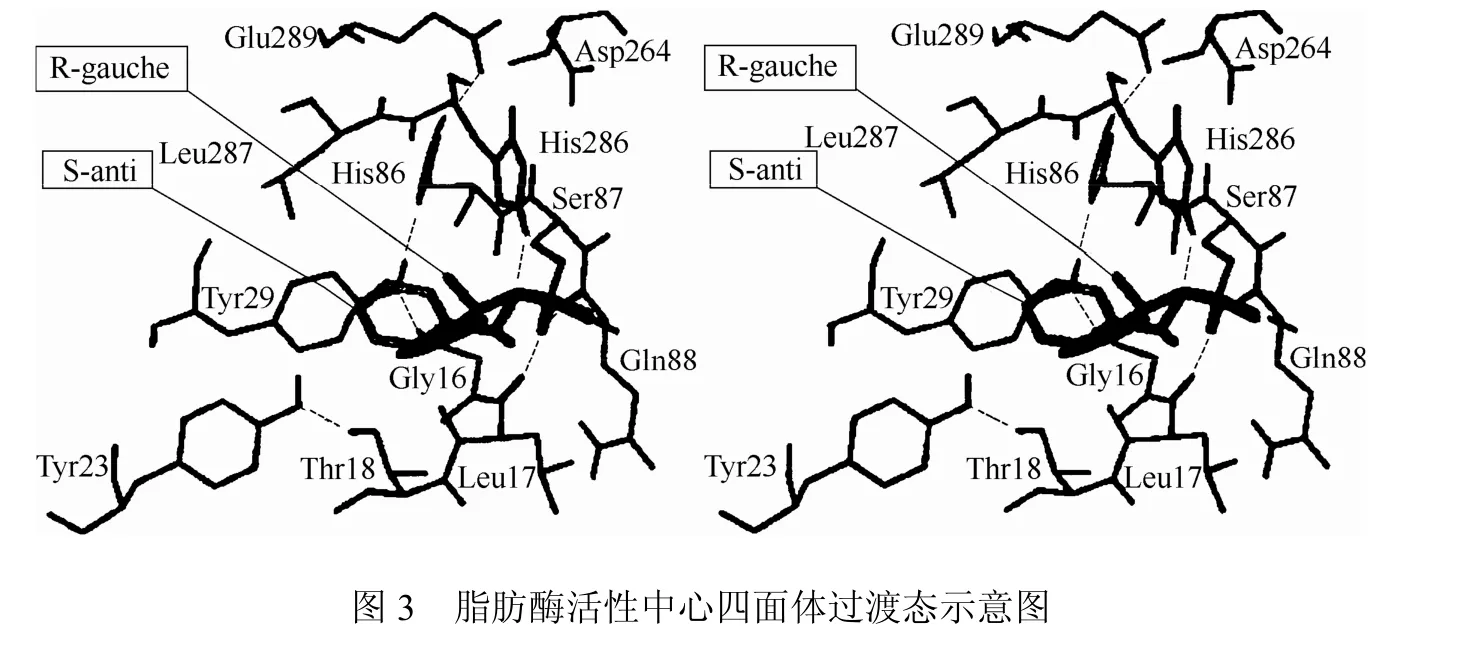

表1 6种脂肪酶水解12种底物的对接打分
已报道的将分子动力学应用于脂肪酶性质和机理研究涉及以下4个方面。①在不同溶剂中的脂肪酶构象变化。Rahman等[14]研究洋葱伯克氏细菌脂肪酶(Burkholderiacepacialipase)在水-辛烷两相混合溶剂体系中构象变化时发现,在水环境中脂肪酶的“盖子”结构处于闭合状态,而当辛烷分子接近“盖子”时,脂肪酶的“盖子”结构打开,且“盖子”打开的幅度与温度有关,温度越高,打开幅度越大。Wedberg等[15]采用分子动力学方法考察了 5种水-有机溶剂混合体系对南极假丝酵母脂肪酶 B(Candida antarcticalipase B)氨基酸残基柔性的影响,得到了类似的结论。②不同 pH 值条件下的酶结构变化。James等[16]将褶皱假丝酵母脂肪酶(Candida rugosalipase)分别置于pH值为5.6和7.2的水溶液中,并进行了2 ns的分子动力学模拟,结果表明:中性pH值条件下脂肪酶“盖子”区的柔性更强,底物更容易进入活性中心区域,因而反应速率更快。③研究脂肪酶与底物间的作用机制和选择性机理[17-20]。Raza等[18]基于分子动力学方法,并通过计算南极假丝酵母脂肪酶 B(Candida antarcticalipase B)与底物形成四面体过渡态后的结合自由能,较好地预测了其对映体选择性。Xu等[19]在研究南极假丝酵母脂肪酶 B与底物的过渡态相互作用过程中发现了几种非常规的复合物结合模式(见图4)。④对脂肪酶的结构进行理性的设计和改造[1,21]。Park等[1]基于分子动力学方法比较了南极假丝酵母脂肪酶B经定点突变前后在有机溶剂中的稳定性,并找到了该脂肪酶中3个可以提高其在有机溶剂中稳定性的关键残基。
对于脂肪酶催化反应来说,首先脂肪酶通过“界面激活”效应打开盖子结构,暴露出催化活性位点,随后底物进入活性区域,与脂肪酶形成四面体过渡态,进而完成催化反应。分子动力学模拟虽然是通过大量的原子运动来优化整个体系,但并没有考虑时间维度,而是把大量动力学模拟产生的构象进行统计,得到合理的平均构象。因而,它无法描述化学键的形成与断裂。


要研究脂肪酶过渡态的成键过程,必须使用量子力学的方法,但由于运算量受到限制,人们尚无法对整个体系进行量子力学计算。为解决这一难题,Warshel等[22]于 1974年首次提出了量子力学/分子力学(quantum mechanics/molecular mechanics,QM/MM)方法,该方法将整个催化体系分为两部分,即精确的量子力学(quantum mechanics,QM)部分和快速的分子力学(molecular mechanics,MM)部分(见图5),采用局部的量子力学研究酶与底物的成键问题,其它理论研究则使用分子力学方法。使用QM/MM方法在一定程度上回避了量子力学计算量过大的问题。众多研究者运用QM/MM方法针对多种脂肪酶的催化机理开展了更加深入的研究[2,23-26](见表 2)。
值得关注的是,近年来人们基于另一种分子模拟方法,即定量构效关系(quantitative structure activity relationships,QSAR)来精确预测脂肪酶的对映体选择性,且预测值与理论值的相关性非常理想(R2> 0.9)[27-28]。QSAR,是运用书写模型来描述分子结构与分子某种生物活性之间的关系。基本假设是化合物的结构包含了其物理、化学以及生物信息,进而决定了该化合物的性质。该方法通过对少量底物的某种生物活性的实际测定,便可直接预测其它同系物的活性。这种方法虽然预测的准确率高,但仅对同系物有效,其适用性不强。
4 存在问题与展望
综上所述,分子对接技术具有快速得到脂肪酶与底物复合物的优势,被广泛应用于高通量预测脂肪酶对底物的选择性研究,计算简单,但准确性较差。而分子动力学方法更接近于真实环境,准确度较高,但其采用的点电荷模型具有不可极化现象和计算过程中无法形成或断开共价键等缺点。QM/MM方法通过局部的量子力学计算,解决了分子动力学方法无法成键的问题,在脂肪酶催化机理研究方面显示出较大优势。目前分子动力学和QM/MM方法仍然存在运算速度慢、预测精确度较差的问题。而QSAR方法虽然预测精确度高,但仅局限于同系物。

表2 QM/MM的方法研究脂肪酶催化机理
随着计算机技术的发展,分子模拟技术在酶的性质及催化机理研究方面的应用将越来越广泛。单一模拟方法已无法满足研究的需要,联合运用多种模拟手段已成为现今研究的主流。当计算机的运算能力逐步提升到不再成为制约模拟方法的关键因素时,根据从头计算的分子动力学方法预测脂肪酶的空间结构、全量子力学研究脂肪酶的催化机理以及新的更加精确的力场,如可极化力场下的分子动力学等将更精确、更接近于真实环境,分子模拟技术将发挥出更加重要的作用。
[1]Park H J,Joo J C,Park K,et al.Prediction of the solvent affecting site and the computational design of stableCandida antarcticalipase B in a hydrophilic organic solvent[J].J.Biotechnol.,2013,163(3):346-352.
[2]Ni Z,Lin X F.Insight into substituent effects in CAL-B catalyzed transesterification by combining experimental and theoretical approaches[J].J.Mol.Model.,2013,19:349-358.
[3]Alder B J,Wainwright T E.Studies in molecular dynamics.1.General method[J].J.Chem.Phys.,1959,31(2):459-466.
[4]Norin M,Haeffner F,Hult K,et al.Molecular-dynamics simulations of an enzyme surrounded by vacuum,water,or a hydrophobic solvent[J].Biophys.J.,1994,67(2):548-559.
[5]Peters G H,vanAalten D M F,Edholm O,et al.Dynamics of proteins in different solvent systems:Analysis of essential motion in lipases[J].Biophys.J.,1996,71(5):2245-2255.
[6]Zuegg J,Honig H,Schrag J D,et al.Selectivity of lipases:Conformational analysis of suggested intermediates in ester hydrolysis of chiral primary and secondary alcohols[J].J.Mol.Catal.B:Enzym.,1997,3:83-98.
[7]Lengauer T,Rarey M.Computational methods for biomolecular docking[J].Curr.Opin.Struc.Biol.,1996,6(3):402-406.
[8]Tafi A,van Almsick A,Corelli F,et al.Computer simulations of enantioselective ester hydrolyses catalyzed byPseudomonas cepacialipase[J].J.Org.Chem.,2000,65(12):3659-3665.
[9]Tyagi S,Pleiss J.Biochemical profiling in silico-predicting substrate specificities of large enzyme families[J].J.Biotechnol.,2006,124(1):108-116.
[10]Xu T,Zhang L J,Wang X D,et al.Structure-based substrate screening for an enzyme[J].BMC Bioinformatics.,2009,10:257-263.
[11]Juhl P B,Doderer K,Hollmann F,et al.Engineering ofCandida antarcticalipase B for hydrolysis of bulky carboxylic acid esters[J].J.Biotechnol.,2010,150(4):474-480.
[12]Piamtongkam R,Duquesne S,Bordes F,et al.Enantioselectivity ofCandida rugosalipases (Lip1,Lip3,and Lip4) towards 2-bromo phenylacetic acid octyl esters controlled by a single amino acid[J].Biotechnol.Bioeng.,2011,108(8):1749-1756.
[13]袁鹏,杨立荣,徐刚,等.共价对接法辅助模拟预测脂肪酶的对映体选择性[J].化工进展,2011,30 (8):1815-1820.
[14]Rahman M Z A,Salleh A,Rahman RNZRA,et al.Unlocking the mystery behind the activation phenomenon of T1 lipase:A molecular dynamics simulations approach[J].Protein Sci.,2012,21(8):1210-1221.
[15]Wedberg R,Abildskov J,Peters G H.Protein dynamics in organic media at varying water activity studied by molecular dynamics simulation[J].J.Phys.Chem.B,2012,116(8):2575-2585.
[16]James J J,Lakshmi B S,Raviprasad V,et al.Insights from molecular dynamics simulations into pH-dependent enantioselective hydrolysis of ibuprofen esters byCandida rugosalipase[J].Protein Eng.,2003,16(12):1017-1024.
[17]Ottosson J,Fransson L,Hult K.Substrate entropy in enzyme enantioselectivity:An experimental and molecular modeling study of a lipase[J].Protein Sci.,2002,11(6):1462-1471.
[18]Raza S,Fransson L,Hult K.Enantioselectivity inCandida antarcticalipase B:A molecular dynamics study[J].Protein Sci.,2001,10(2):329-338.
[19]Xu T,Zhang L J,Su E Z,et al.Disparity in productive binding mode of the slow-reacting enantiomer determines the novel catalytic behavior ofCandida antarcticalipase B[J].J.Mol.Catal.B:Enzym.,2010,62:288-296.
[20]Yagnik A T,Littlechild J A,Turner N J.Molecular modelling studies of substrate binding to the lipase fromRhizomucor miehei[J].J.Comput.Aid.Mol.Des.,1997,11(3):256-264.
[21]Maraite A,Hoyos P,Carballeira J D,et al.Lipase fromPseudomonas stutzeri:Purification,homology modelling and rational explanation of the substrate binding mode[J].J.Mol.Catal.B:Enzym.,2013,87:88-98.
[22]Warshel A,Levitt M.Theoretical studies of enzymic reactions:Dielectric,electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme[J].J.Mol.Biol.,1976,103(2):227-249.
[23]Luic M,Stefanic Z,Ceilinger I,et al.Combined X-ray diffraction and QM/MM study of theBurkholderia cepacialipase-catalyzed secondary alcohol esterification[J].J.Phys.Chem.B,2008,112:4876-4883.
[24]Tosa M,Pilbak S,Moldovan P,et al.Lipase-catalyzed kinetic resolution of racemic 1-heteroarylethanols-experimental and QM/MM study[J].Tetrahedron-Asymmetry,2008,19:1844-1852.
[25]Bellucci L,Laino T,Tafi A,et al.Metadynamics simulations of enantioselective acylation give insights into the catalytic mechanism ofBurkholderia cepacialipase[J].J.Chem.Theory.Comput.,2010,6:1145-1156.
[26]Brem J,Pilbak S,Paizs C,et al.Lipase-catalyzed kinetic resolutions of racemic 1-(10-ethyl-10H-phenothiazin-1,2,and 4-yl)ethanols and their acetates[J].Tetrahedron-Asymmetry,2011,22:916-923.
[27]Wang H K,Wang X J,Li X L,et al.QSAR study and the hydrolysis activity prediction of three alkaline lipases from different lipase-producing microorganisms[J].Lipids Health Dis.,2012,11:124-132.
[28]Gu J L,Liu J,Yu H W.Quantitative prediction of enantioselectivity ofCandida antarcticalipase B by combining docking simulations and quantitative structure-activity relationship (QSAR) analysis[J].J.Mol.Catal.B:Enzym.,2011,72:238-247.
