质子交换膜燃料电池膜电极制备方法及部分改进策略
2013-10-11郑俊生马建新
田 甜 ,郑俊生 ,马建新 ,3
(1华东理工大学资源与环境工程学院,上海 200237;2同济大学(嘉定校区)新能源汽车工程中心,上海 201804;3同济大学(嘉定校区)汽车学院,上海 201804)
燃料电池是通过电化学反应将储存在反应物中的化学能直接转化为电能的能量转换装置,其中不涉及热机的燃烧过程。燃料电池由于具有环境友好、能量密度高、室温下可快速启动和可靠性高等优点[1],有望替代内燃机而应用在汽车领域。美国通用汽车公司(General Motors,GM)已经开发了几代燃料电池汽车,电池的Pt使用量已由第四代的0.85 g/kW降到第五代的0.32 g/kW,计划到2015年将Pt使用量再进一步降低到0.11 g/kW以下,并实现第五代燃料电池汽车的商业化[2]。
与其它燃料电池相比,质子交换膜燃料电池(proton exchange membrane fuel cell,PEMFC)的工作温度相对较低,适合用作电动车载、便携式和住宅用电源[3]。膜电极(membrane electrode assembly,MEA)是PEMFC的核心部件,它是电化学反应发生的重要场所,主要由气体扩散层(gas diffusion layers,GDLs)、催化剂层(catalyst layers,CLs)和膜组成。由于电化学反应的发生需要各个组成部分的协调配合,所以膜电极的制备方法、组装工艺、物化特性、使用材料和运行条件等都会对PEMFC的性能产生重要影响。成本和寿命问题一直是制约PEMFC实现商业化的瓶颈,研究者们力求在不影响电池性能的前提下,通过降低催化剂使用量,优化膜电极制备工艺来进一步拓宽PEMFC的应用范围。
本文以提高PEMFC性能为出发点,综述了有关MEA组成部分和运行条件优化的研究进展。
1 膜电极的制备方法
根据MEA制备过程中催化剂层支撑基体的不同,常将膜电极的制备方法分为催化剂制备到基体上(catalyst-coated substrate,CCS)和催化剂制备到膜上(catalyst-coated membrane,CCM)法两种。CCS法是以GDLs作为催化剂层的支撑基体,先混合催化剂及溶剂制备催化剂浆液,然后采用不同方法将催化剂浆液应用到经过预处理的碳纸或碳布上,制得多孔气体扩散电极,最后将制得的多孔气体扩散电极与处理过的膜材料热压形成MEA。而CCM法通常采用质子交换膜(proton exchange membrane,PEM)作为催化剂层的支撑基体,采用不同方法使催化剂层直接加到膜上形成MEA,使用时再在MEA两侧应用GDLs即可。基于CCS法制备电极技术的优势在于不涉及“膜吸水”褶皱问题,所以催化剂浆料配比要求相对较低,可选用喷涂、刷涂、刮涂、丝网印刷、拉浆等多种方法进行,因而设备及工艺相对较为成熟而简单。CCS法的缺点是容易导致催化剂浆料渗透扩散层中,造成催化剂利用率低、改变扩散层的孔结构和憎水性,增加传质浓差极化和欧姆电阻;另一方面,CCS法电极容易导致电极与膜之间离子传导结合不佳,界面电阻大。为了克服CCS法的缺陷,进一步提高催化剂利用率,降低贵金属载量,人们提出了CCM电极制备方法。
CCS和CCM法中常见的MEA制备方法有溅射法、喷涂法、转印法、浸渍还原法和丝网印刷法。下文就溅射法、喷涂法和转印法作简单的介绍。
1.1 溅射法
通过溅射法可以制得均匀分布的催化剂层[4],制备过程中,Pt纳米颗粒能够有效地沉积在三相界面,在一定程度上提高了Pt利用率。在真空条件下制备还能得到低Pt含量的催化电极[5]。另外,溅射沉积能够简化电极的制备过程,降低制造成本[6]。溅射法的一般流程是:在真空和室温下,调节溅射装置参数,将催化剂浆液加到处理过的膜或GDLs上,再组装成MEA[7-8]。Hayre等[6]采用真空溅射法制备了CCM膜电极,将经过 H2SO4、H2O2和去离子水处理后的Nafion膜置于真空室内,在氩气的溅射气压为0.67 Pa、体积流量为30 sccm的条件下,先将Pt溅射沉积在膜的一面,然后从真空室中取出Nafion膜,翻面后再送入真空室继续溅射沉积。
Lai等[9]采用RF(Radio Frequency)磁控溅射方法先在GDLs上沉积不同负载量(0.02 mg/cm2、0.04 mg/cm2、0.10 mg/cm2、0.20 mg/cm2、0.40 mg/cm2)的Pt催化剂,经刷涂 Naf i on溶液后再与Naf i on117热压形成MEA。考察了阴极Pt负载量对电极性能的影响,表征结果得到Pt负载量为0.1 mg/cm2时的极化电阻最低,电池性能也优于较高Pt负载量(0.2、0.4 mg/cm2)的。通过这种方法沉积的Pt催化剂利用率优于其它不经溅射方法得到的电极[10]。
但溅射法制备的MEA的耐久性问题还有待解决[11],且溅射过程中会产生催化剂的流失问题[6,11]。
1.2 喷涂法
由于操作简便、仪器简单,实验室常用喷涂法来制备电极。喷涂法的一般流程是:先制备合适的催化剂浆液,然后用喷枪设备将催化剂浆液喷涂到GDLs或经处理过的膜两侧,在一定条件下干燥一段时间后祛除多余杂质,再与膜或GDLs组装成MEA[12-13]。André Wolz 等[14]利用喷涂法制备了一种多层结构的膜电极。整个喷涂过程在120℃条件下(以加速干燥过程)进行,先将 Nafion膜固定在与喷雾锥相垂直的方向上,然后用Ecospray喷涂装置将含Pt/PANI(聚苯胺)的催化剂浆液涂到膜的一侧,持续4 s,然后停留2 s使涂层完全干燥,接着再喷涂含Pt/CNTs的催化剂浆液,同样持续4 s,停留2 s使涂层完全干燥,如此反复喷涂直到浆液用完。在膜的另一侧,用相同的喷涂装置涂上含Pt/C的催化剂浆液,即制得CCM型MEA。
Koraishy等[15]认为喷涂参数与喷嘴特性能够影响催化剂浆液形成的液滴大小,液滴大小改变电极结构从而使MEA的性能发生变化。Chaparro等[16]发现电喷涂方法能够引起严重的质量传输损失及电催化剂的部分团聚。在低电流密度下,电喷涂法制备的膜电极(Pt负载量0.3 mg/cm2)稍优于Pt负载量为0.5 mg/cm2时的电极和商业E-TEK电极,但在高电流密度下由于H+或氧气的传输极化,电极性能会下降。同时,喷涂法会引起催化剂和高聚物发生团聚现象[17]。
1.3 转印法
转印法[18]被认为是实现MEA大规模生产的最简便方法。转印法中,先制备包含Pt催化剂、高分子离聚物和溶剂的催化剂浆液,常用刮墨刀片法(doctor blade)将浆液涂覆在转印基质上,使溶液蒸发以形成三相界面,然后同阴阳极和膜热压,再祛除转印基质[19-20]。
Saha等[21]采用一种基于胶体浆液的改进转印方法来制备CCM膜电极,先将制备好的催化剂浆液喷洒到Tef l on sheet上,在120℃下干燥2 h后,采用热压法将催化剂层转移到经预处理的H+型Naf i on112膜上,形成CCM,再在CCM的阴极和阳极侧加上气体扩散层制得膜电极。与传统的基于溶液浆液的方法(solution ink decal method)相比,这种改进方法制备的电极具有更多的孔结构和更大的Pt/C团聚物,电化学表面积也更高。基于这些优势,在大电流密度下的质量传输性能得以提高,900 mV时的电流密度为15.5 mA/cm2(vs 6.3 mA/cm2),600 mV时的功率密度为 0.38 W/cm2(vs 0.22 W/cm2)。
CCM法中,直接将湿浆液喷涂到膜上时,可能会引起膜的收缩或膨胀问题。采用转印方法能够避免这种问题的发生[18]。但是,Cho等[20]认为转印法还存在以下几个问题:①催化剂不能完全地从转印基质转移到膜上;②高电流密度下的质量传输受阻问题;③催化剂浆液分散不均匀,影响Pt的利用率和质子电导率。
2 MEA制备方法的改进
CCS法和CCM法已普遍应用于实际生产与实验室研究中。但基于这两种方法制备的MEA还存在一些缺陷,比如多孔气体扩散电极通常含有一定量的聚四氟乙烯(PTFE)憎水剂,这样做在一定程度上能够促进反应气体的扩散传输,但不利于电子的传导;催化剂被离子聚合物包覆不能充分地参与电化学反应,这部分催化剂得不到利用,降低了催化剂利用率;随着电池运行时间的延长,CLs容易从PEM表面脱落,影响电极使用寿命等。这些都严重地影响电极的性能与使用寿命。在此基础上,通过改进制备方法、优化制备条件、选取合适材料,还能够进一步提高MEA性能。
2.1 Nafion含量的优化
在电极制备过程中,常常以活性金属和Nafion作为制备催化剂层的原料,其中Nafion含量对所制备电极的性能有很大影响[22]。一定量的Nafion虽然能够促进质子的传导,但过多的Nafion一方面使得电极产生“水淹”现象,气体传输阻力增大,表现出明显的传质极化损失;另一方面,降低电极的孔隙率,包裹催化剂,降低催化剂利用率。但是,Nafion具有一定的粘附作用,能够使催化剂层与膜产生良好的接触而形成三相反应界面。因此,优化Nafion含量对电化学反应的有效发生至关重要。
Xie等[23]考察了阴极催化剂层中的Nafion含量对MEA结构和性能的影响。当Nafion质量分数大于28%时,MEA性能随着Nafion含量的减少而提高;当Nafion含量小于28%时,MEA性能则随着Nafion含量的减少而降低,尤其在质量传输区域。Nafion含量对整个极化区域都有重要影响,活化区域(0.9 V和0.8 V)下,电流密度随着Nafion含量的增加先缓慢增加后逐渐减小;欧姆(0.7 V和0.6 V)和质量传输区域(0.4 V和0.3 V)下,电流密度随着Nafion含量的增加先缓慢增加后急剧减小。这些结果表明了阴极催化剂层中的Nafion含量能够影响OOR(oxygen reduction reaction)动力学、电荷及不带电物质的传输扩散。以E-TEK 20%Pt3Cr/C作阴极催化剂时,Nafion的最佳含量为(27±6)%。
2.2 质子交换膜的改进
氟化聚合物膜为目前PEMFC最常用的质子交换膜材料,质子交换膜的质子导电性、厚度、化学稳定性、力学性能能够影响电池寿命和电极性能。
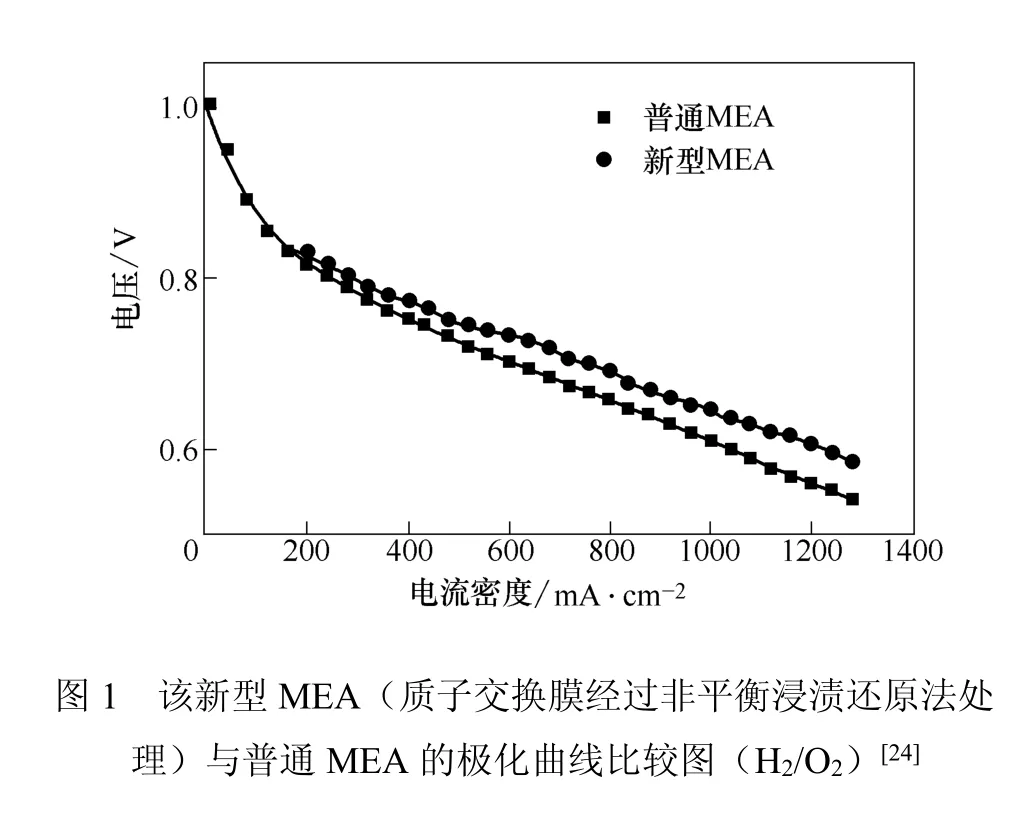
Pethaiah等[24]采用非平衡浸渍还原法制备了一种新型MEA并同普通MEA进行了比较,两种MEA都采用相同的CCS法制备气体扩散电极,但是对质子交换膜的处理方式有所差别。普通MEA的质子交换膜未经非平衡浸渍还原处理,新型MEA的质子交换膜处理过程如下:Naf i on 1135膜依次经5%的H2O2、Millipore水、0.5 mol/L H2SO4和高纯水除去杂质后,以0.5 mol/L NaCl处理转化为Na+型膜,然后将这种Na+型Naf i on膜浸渍在甲醇、Pt(NH3)4C12和水的混合溶液中,调节溶液pH值到13,加入还原剂10 mmol/L NaBH4后搅拌2 h。以同样方法处理该质子交换膜的另一面,经0.5 mol/L H2SO4和去离子水洗后,即制得Pt/PEM/Pt型膜,最后将该膜与气体扩散电极热压形成MEA。两种MEA的极化曲线如图1所示。
这种方法制得的膜电极具有比普通MEA更大的电化学表面积和粗糙度,三相界面扩大,Pt的利用率因此得到提高,在一定程度上可以减低电池成本。另外,质子交换膜的稳定性也得到了改善。
2.3 热压条件的优化
热压过程是组装两电极和质子交换膜的一种简便方法,常用在CCS法中。热压过程的工作温度一般在130℃左右,在这一过程中,MEA的内部结构、孔隙率和性能均会发生改变[25]。随着热压压力的增加,催化剂层的离子电导率和三相反应界面面积先增加后减小。热压压力低时,电极和质子交换膜发生分离。热压时间不能过长,否则引起电极孔隙率减小,质量传输阻力增大。另外,高温使得Naf i on膜失水,高分子离聚物降解,电极和膜发生部分分离[20-21,26-27]。
Yazdanpour等[28]先用喷墨印刷法(IJP Method)制得了 Pt/MWCNTs(Multi-walled Carbon Nanotubes)型MEA,发现经过热压过程的MEA性能优于不经热压过程的。同时,考察了热压参数对MEA性能的影响,得到在800 psi、100℃和3 min的条件下,该Pt/MWCNTs型MEA的功率密度最高。Taguchi experimental分析结果显示了热压压力对MEA的影响最大,其次是热压温度,最后是热压时间。
2.4 催化剂层的改进
通过制备一种带梯度结构的催化剂层,可以很好地平衡贵金属使用量与电极性能的关系。在靠近质子交换膜的一端,Pt负载量和Nafion含量较高,增加了与电解质的接触面积,缩短了质子传输路径;而在CLs与GDLs的界面,孔径较大,Nafion含量较少,孔不容易被Nafion覆盖,促进了反应气的扩散和水的排除,使欧姆电阻降低。
Su等[26]采用CCM法来制备了超低Pt载量的膜电极组件,这种MEA带有两层催化剂层,内层Pt负载量和Naf i on含量均较高以确保催化剂利用率,外层Pt负载量和Naf i on含量均较低以得到厚度适宜的MEA,从而促进催化剂层中的质量传输过程。分别将含有40%Pt/C和10%Pt/C的浆液喷涂在Naf i on 212膜的两面形成内外催化剂层,内外层的厚度一致。结果表明,这种新型双催化剂层膜电极的性能大大提高,尤其是在高电流密度下。0.65 V下,当阴阳极Pt负载量分别为0.12 mg/cm2和0.04 mg/cm2时,它的电流密度可以达到0.73A/cm2。在1.3A/cm2和0.51 V下,它的最大功率密度达到0.66 W/cm2,比单催化剂层膜电极高11.9%,电化学阻抗谱(electrochemical impedance spectroscopy,EIS)和循环伏安法(cyclic voltammetry,CV)结果表明,这种新型双催化剂层膜电极具有有效的电化学活化层及良好的Pt利用率。
在过去的十多年里,3M公司开发了继Pt黑和分散型Pt/碳黑之后的第三代燃料电池电催化剂——纳米结构薄膜(nano-structured thin film,NSTF)催化剂[29]。这种催化剂采用呈矩形条状、纵横比较高的有机晶须(Whiskers)作为载体[30-31],以溅射法[29-30,32]将催化剂沉积到上面。纳米结构薄膜电催化剂在改善车用燃料电池的使用寿命、性能和降低成本方面具有良好的发展前景。它的比活性、稳定性和使用寿命远远高于传统的高分散Pt/C催化剂[30,32]。
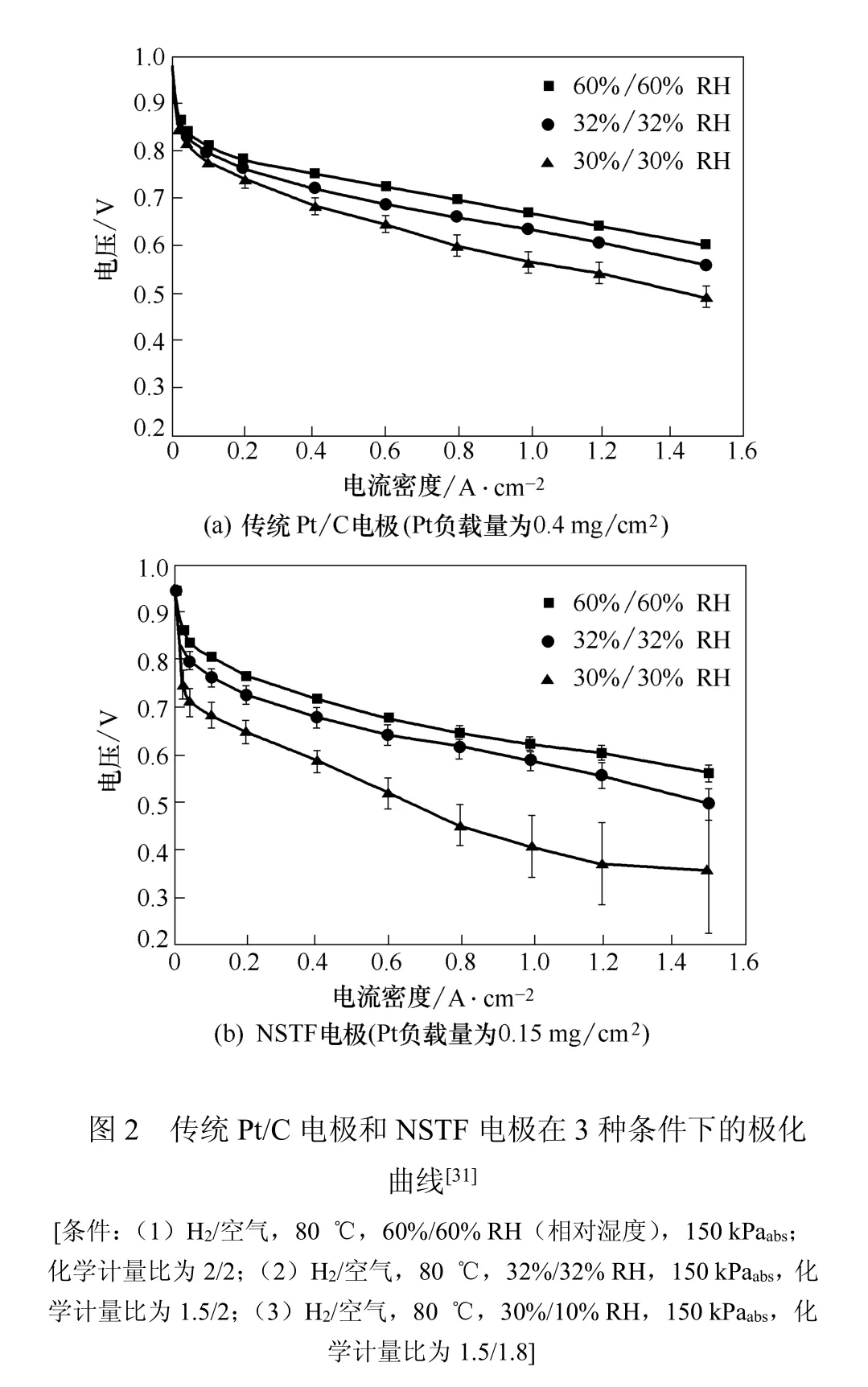
3M 公司制备的NSTF MEA基于卷挠(rollgood)CCM法[33],不含碳和高分子离聚物,厚度仅为传统Pt/C MEAs的1/30~1/20[34]。Sinha等[31]将NSTF PtCoMn催化剂层压到PFSA膜上,层压过程中NSTF晶须(NSTF whiskers)压缩使电极变薄(≤0.23 mm),部分晶须渗入膜内,其余晶须不添加任何高聚物,并比较了NSTF MEA与传统Pt/C MEA的性能。极化曲线如图2(a)和2(b)所示。
实验发现在80℃,较高相对湿度(RH)条件下,Pt负载量为0.15 mgPt/cm2的NSTF电极性能同Pt负载量为0.40 mgPt/cm2的传统Pt/C电极相当。但在干燥条件下,NSTF电极的质子电导率不佳从而影响了Pt的利用率,使电极性能下降。因此他们认为应该着重研究NSTF的水管理问题从而提高电极性能。
载体材料能够影响催化剂的利用率、分散性和孔径分布等性能。碳黑(carbon blacks),尤其是Vulcan XC-72,是最常用的Pt和Pt合金催化剂载体材料[35]。XC-72C[36]虽然具有高表面积(250 m2/g),可以提高催化剂的分散性。但是,它通常含有许多微孔(直径<2 nm),使溶剂化的离子难以通过且易被Naf i on覆盖,导致催化剂的利用率降低。另外,它含有的硫会引起Pt颗粒的团聚,使Pt颗粒粒径增大。
碳纳米管(carbon nanotubes,CNTs)由于导电性强、化学稳定性好、表面积大等优点[37],已有众多报道研究[38-40]了其作为催化剂载体材料来使用。与XC-72相比,纳米管的形态结构更倾向于分散电催化剂而不是使它们相互结合,当阳极引入含CO的H2时,CNTs基催化剂的性能也更好[41]。CNTs是一种呈化学惰性的特殊的高(大)分子,当它的结构发生改变或者表面出现一些活性官能团[42-43](如羟基、羰基)时,才有可能发生化学反应[39,44]。Huang等[43]发现含CNTs的电极性能也优于不含CNTs的,以CNTs作为第二载体可以提高与催化剂的接触表面积,加速ORR。
Yan等[45]制备了一种Pt/CNTs/CP MEA,制备过程包括以下几个步骤。①在CP(Carbon Paper)上生长 CNTs。在Co/Ni合金催化剂的作用下,通过化学气相沉积法将MCNTs直接沉积到CP的电活性位上。②CNTs的表面功能化和Pt的沉积。在三电极体系中,以CNTs/CP样品为工作电极,4-甲氧基苯基重氮四氟硼酸盐和四丁基四氟硼酸铵的乙腈溶液(4-methoxyphenyldiazonium tetrabutylammonium tetrafluoroborate acetonitrile)为电解质,通过电化学还原法使CNTs表面带有苯基基团,然后用98%H2SO4溶液处理以引入磺酸基,接着将样品浸到H2PtCl6水溶液中,使Pt4+吸附到磺酸基团上。最后,在600℃、H2条件下,Pt4+还原成Pt,同时功能基团移开CNTs表面。③Pt/CNTs/CP MEA的制备。先用PTFE、炭黑的异丙醇溶液制备气体扩散浆液,然后将该浆液涂到Pt/CNTs/CP电极的CP侧,在340℃下煅烧1 h后,再在CNTs侧喷上含2.5%(质量分数)Nafion的异丙醇溶液。最后在 140℃下煅烧1 h,即制得Pt/CNTs/CPMEA。
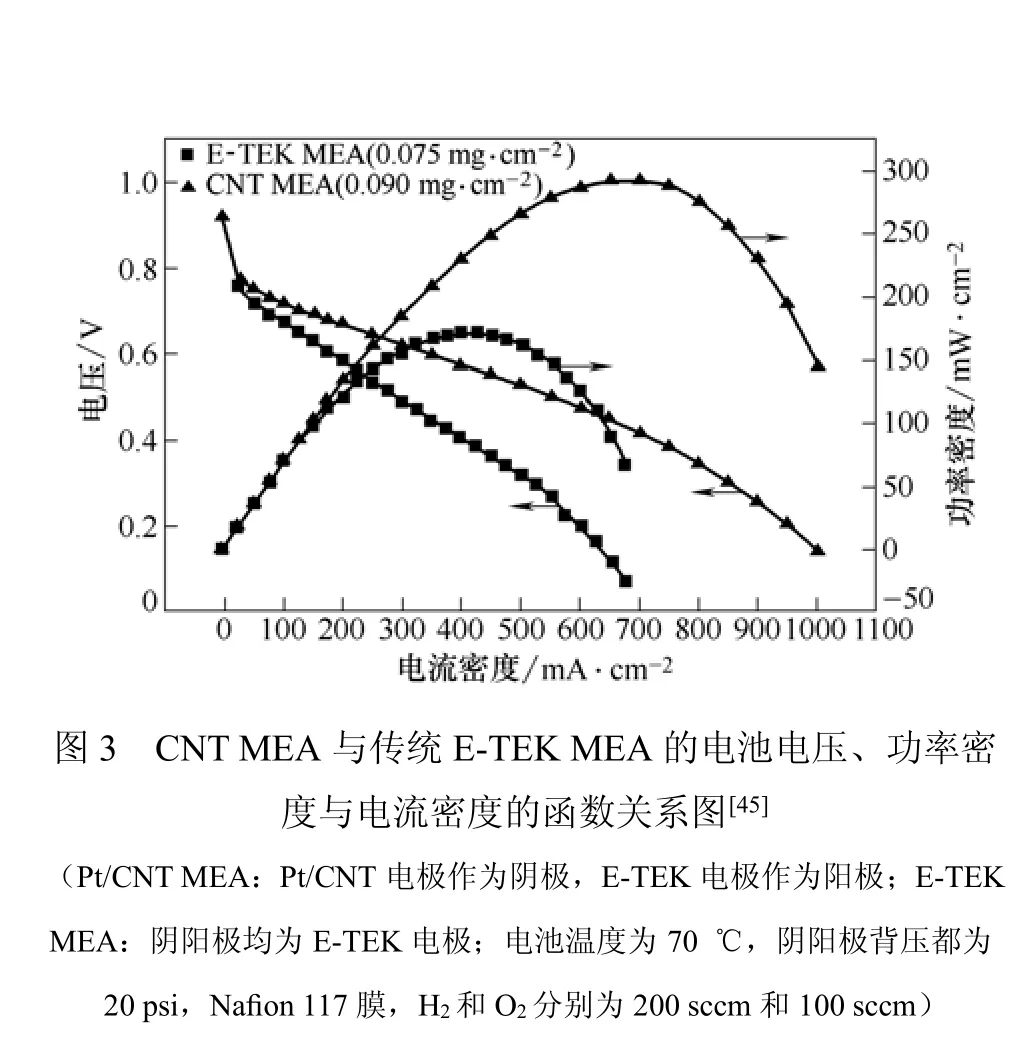
这种MEA(阴极Pt载量为0.09 mg/cm2)的比表面积、Pt利用率、阴极比活性和电极性能均优于传统的E-TEK MEA(阴极Pt载量为0.075 mg/cm2),极化曲线如图3所示。
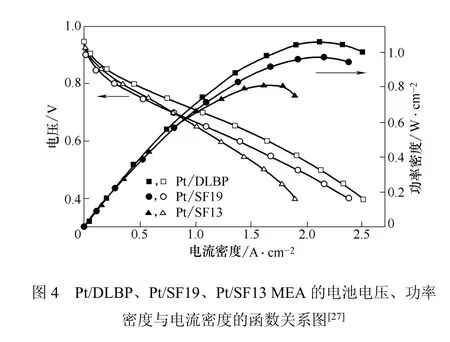
Zheng等[27]制备了一种带有梯度结构的双层巴克纸(Buckypaper,BP)电极,催化剂层中的孔隙率、Pt负载量和Nafion含量沿BP的厚度呈梯度分布,BP载体包括SCNTs+CNFs子层和CNFs子层。采用这种Pt/DLBP作为阴极制得的MEA在相对较低的Pt载量下具有较高的功率。当阴极Pt载量为0.11 mg/cm2,电压为0.65 V时,可以取得0.88 W/cm2的额定功率和0.18 gPt/kW的总Pt利用率(高于目前传统Pt/C催化剂取得的最佳值0.4 gPt/kW)。另外,由于CNFs的高度石墨化和优良的耐腐蚀性,该Pt/DLBP电极具有很高的稳定性。极化曲线如图4所示。
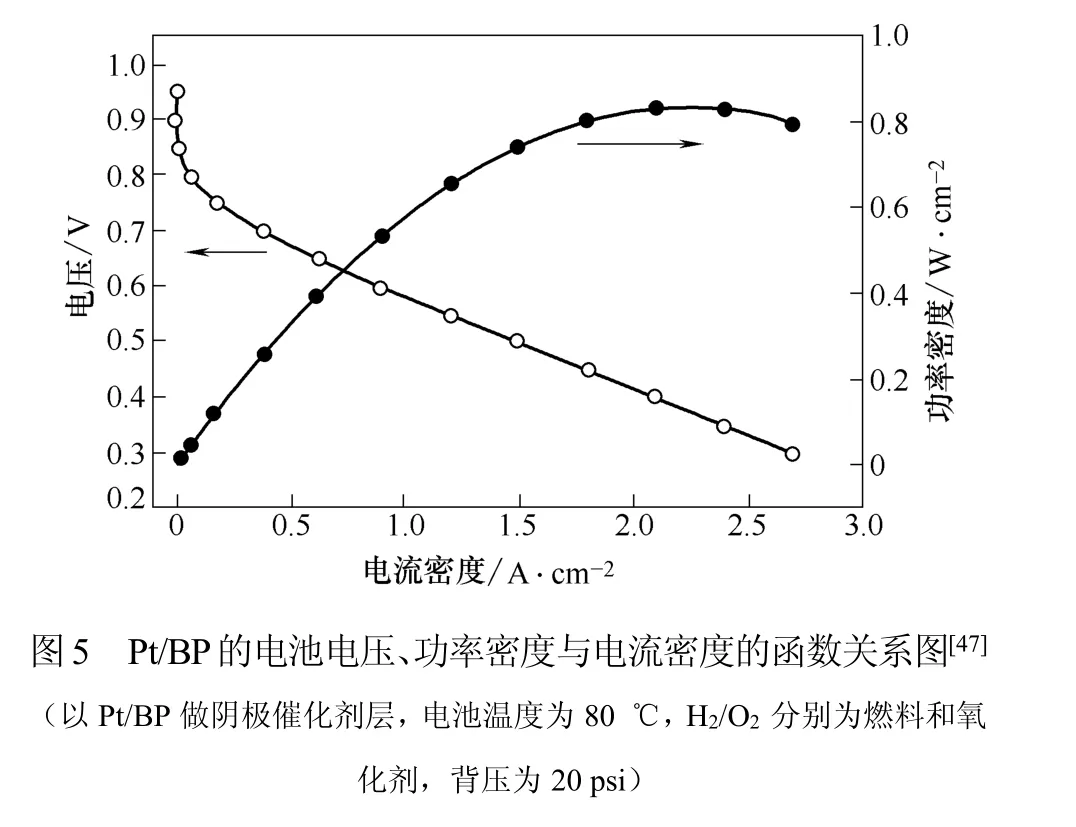
使用不同的沉积方法可以得到不同结构、尺寸和形态的活性金属颗粒及粒径分布。目前,活性金属的沉积普遍存在难以均匀沉积且粒径较大的问题。Zheng等[46]采用脉冲电沉积方法在BP上沉积Pt,得到了平均粒径为5~6 nm的催化剂颗粒。为进一步降低颗粒尺寸,Zheng等[47]运用超零界流体法制备了另一种超低Pt载量电催化剂。超零界流体制备过程如下:将功能化的BP与一定量的PtMe2COD前体溶液加到容器中,在70℃加热后,容器被二氧化碳缓慢地加压到3500 psi,保持这个条件6 h使溶液达到平衡状态。然后冷却称量浸渍过的BP,再在管式炉中加热4 h(N2,200℃)使Pt2+还原为Pt。最终制得的Pt颗粒粒径为2~4 nm。极化曲线如图5所示:以此催化剂组装的MEA在超低Pt载量的情况下仍具有很高的Pt利用率。当Pt载量为0.025 mg/cm2,电压为0.65 V时,可以取得0.4 W/cm2的功率和0.06 gpt/kW的阴极Pt利用率。
Kim等[48]采用一种新型的电流替代法(galvanic replacement process)制备了PtMONC纳米复合催化剂。该方法不同于传统的金属沉积方法,不需要添加任何还原物质,具体的制备过程如下:70℃下,向Sf-MON(不含表面活性剂的Mn3O4纳米粒子)的水溶液中加入Na2PtCl4溶液,搅拌2 h。搅拌过程中,Mn3O4与Na2PtCl4发生反应,在Sf-MON球形表面形成密集均匀的Pt纳米晶体,冷却到室温后,通过离心和去杂质得到PtMONC。反应时间为30 min时,沉积得到的Pt粒径为1.3~1.9 nm。反应时间为4 h时,沉积得到的Pt粒径为1.9~2.7 nm。这种PtMONC纳米催化剂的比活性和耐久性均优于商业Pt/C催化剂。比活性和耐久性分别如图6和表1所示。
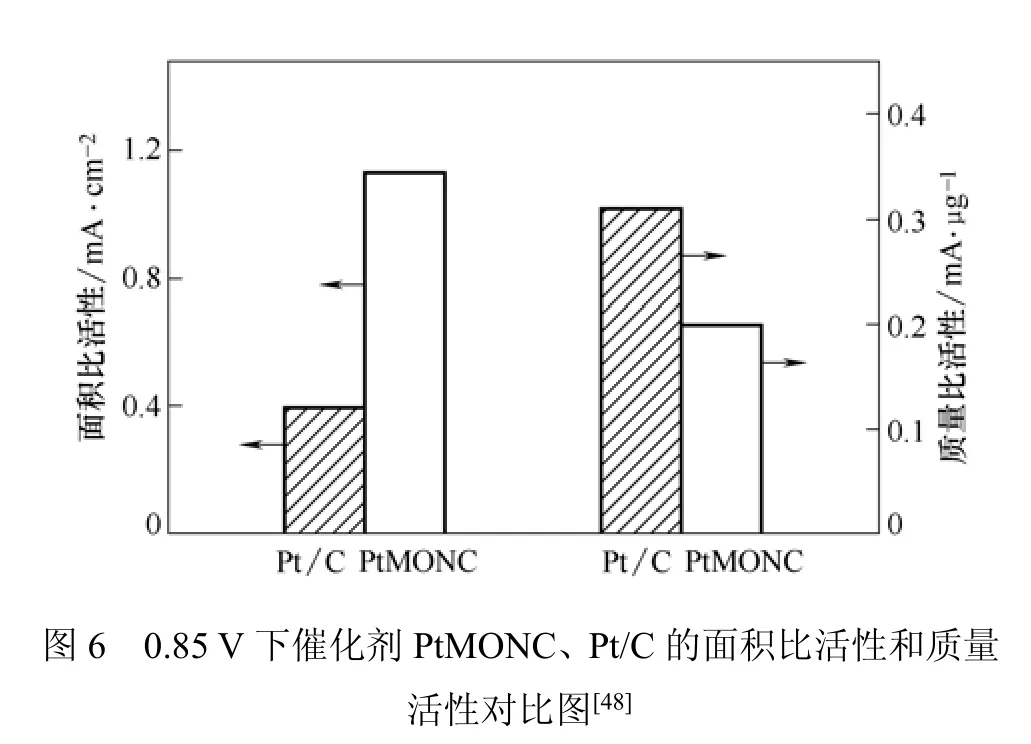
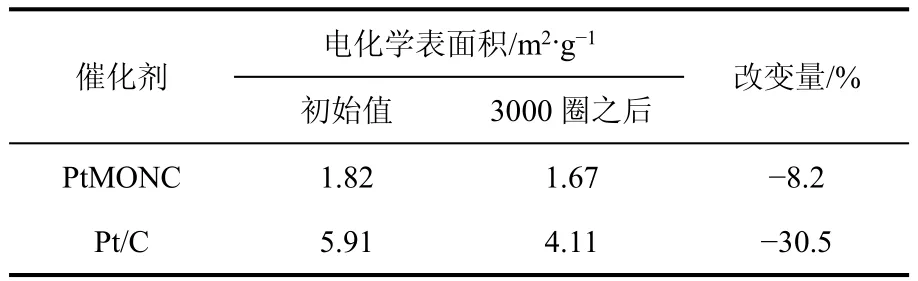
表1 PtMONC与Pt/C的电化学表面积变化[48]
3 结论与展望
为了解决CCS法中存在的不足,提出了CCM法,并通过优化离子聚合物含量和热压条件、对质子交换膜进行处理以及改进催化剂层结构,来寻求优化的膜电极制备工艺。
很多研究者从梯度结构、NSTF、碳纸上原位生长碳纳米管、碳纳米管/碳纳米纤维复合网状物等方面来改进催化剂层结构。通过喷涂不同活性金属和高聚物含量的浆液,可以制得呈梯度分布的催化剂层,然后运用性能优良的材料作为载体层,在一定条件下,制得的膜电极性能优于传统的Pt/C或E-TEK电极。
[1]Hayre R O,车硕源,Colella W等著.燃料电池基础[M].王晓红,黄宏,等译.北京:电子工业出版社,2007.
[2]Sam Abuelsamid.Honey,I shrunk the fuel cell!Next-gen GM hydrogen stack gets small[OL].[2009-08-17].http://green.autoblog.com/2009/08/17/honey-i-shrunk-the-fuel-cell-next-gen-gm-hydrogenstack-gets-s/.
[3]Nishikawa O,Doyama K,Miyatake K,et al.Gas diffusion electrodes for polymer electrolyte fuel cells using novel organic/inorganic hybrid electrolytes:Effect of carbon black addition in the catalyst layer[J].Journal of Electrochimica Acta,2005,50(13),2719-2723.
[4]Natarajan S K,Hamelin J.High-performance anode for polymer electrolyte membrane fuelcellsby multiple layerPtsputter deposition[J].Journal of Power Sources,2010,195(22):7574-7577.
[5]Huang K L,Lai Y C,Tsai C H.Effects of sputtering parameters on the performance of electrodes fabricated for proton exchange membrane fuel cells[J].Journal of Power Sources,2006,156(2):224-231.
[6]Hayre R O,Lee S J,Cha S W,et al.Asharp peak in the performance of sputtered platinum fuel cells at ultra-low platinum loading[J].Journal of Power Sources,2002,109(2):483-493.
[7]Caillard A,Brault P,Mathias J,et al.Deposition and diffusion of platinum nanoparticlesin porous carbon assisted by plasma sputtering[J].Surface and Coatings Technology,2005,200(1-4):391-394.
[8]Rabat H,Brault P.Plasma sputtering deposition of PEMFC porous carbon platinum electrodes[J].Fuel Cells,2008,8(2):81-86.
[9]Lai Y C,Huang K L,Tsai C H,et al.Sputtered Pt loadings of membrane electrode assemblies in proton exchange membrane fuel cells[J].International Journal of Energy Research,2012,36(8):918-927.
[10]Gruber D,Ponath N,Müller J,et al.Sputter-deposited ultra-low catalyst loadings for PEM fuel cells[J].Journal of Power Sources,2005,150:67-72.
[11]Cho Y H,Yoo S J,Cho Y H,et al.Enhanced performance and improved interfacial properties of polymer electrolyte membrane fuel cells fabricated using sputter-deposited Pt thin layers[J].Electrochimica Acta,2008,53(21):6111-6116.
[12]Thanasilp S,Hunsom M.Effect of MEA fabrication techniques on the cell performance of Pt-Pd/C electrocatalyst for oxygen reduction in PEM fuel cell[J].Fuel,2010,89(12):3847-3852.
[13]Sun L L,Ran R,Wang G X,et al.Fabrication and performance test of a catalyst-coated membrane from direct spray deposition[J].Solid State Ionics,2008,179(21-26):960-965.
[14]WolzA,Zils S,Michel M,et al.Structured multilayered electrodes ofproton/electron conducting polymerforpolymerelectrolyte membrane fuel cells assembled by spray coating[J].Journal of Power Sources,2010,195(24):8162-8167.
[15]Koraishy B M,Meyers J P,Wood K L.Manufacturing of Direct Methanol Fuel Cell Electrodes by Spraying[J].Journal of the Electrochemical Society,2011,158(12):B1459-B1471.
[16]Chaparro A M,Ben’ıtez R,Gubler L,et al.Study of membrane electrode assemblies for PEMFC With cathodes prepared by the electrospay method[J].Journal of Power Source,2007,169(1):77-84.
[17]Hwang D S,Park C H,Yi S C,et al.Optimal catalyst layer structure of polymer electrolyte membrane fuel cell[J].International Journal of Hydrogen Energy,2011,36(16):9876-9885.
[18]Krishnan N N,Prabhuram J,Hong Y T,et al.Fabrication of MEA with hydrocarbon based membranes using low temperature decal method for DMFC[J].International Journal of Hydrogen Energy,2010,35(11):5647-5655.
[19]Kim K H,Lee K Y,Kim H J,et al.The effects of Naf i on ionomer content in PEMFC MEAs prepared by a catalyst-coated membrane(CCM)spraying method[J].International Journal of Hydrogen Energy,2010,35(5),2119-2126.
[20]Cho H J,Jang H,Lim S,et al.Development of a novel decal transfer process for fabrication of high performance and reliable membrane electrodeassembliesforPEMFCs[J].InternationalJournalof Hydrogen Energy,2011,36(19):12465-12473.
[21]Saha M S,Paul D K,Peppley B A,et al.Fabrication of catalyst-coated membrane by modi fi ed decal transfer technique[J].Electrochemistry Communications,2010,12(3):410-413.
[22]王晓丽.质子交换膜燃料电池膜电极结构研究[D].北京:中国科学院研究生院,2006.
[23]Xie J,Xu F,Wood III D L,et al.Influence of ionomer content on the structure and performance of PEMFC membrane electrode assemblies[J].Electrochimica Acta,2010,55(24):7404-7412.
[24]Pethaiah S S,Kalaignan G P,Sasikumar G,et al.Evaluation of platinum catalyzed MEAs for PEM fuel cell applications[J].Solid State Ionics,2011,190(1):88-92.
[25]Song C,Pickup P G.Effect of hot pressing on the performance of direct methanol fuel cells[J].Journal of Applied Electrochemistry,2004,34(10):1065-1070.
[26]Su H N,Zeng Q,Liao S J,et al.High performance membrane electrode assembly with ultra-low platinum loading prepared by a novel multi catalyst layer technique[J].International Journal of Hydrogen Energy,2010,35(19):10430-10436.
[27]Zhu W,Zheng J P,Liang R,et al.Ultra-low platinum loading high performance PEMFCs using buckypaper-supported electrodes[J].Electrochemistry Communications,2010,12(11):1654-1657.
[28]Yazdanpour M,Esmaeilifar A,Rowshanzamir S.Effects of hot pressing conditions on the performance of Nafion membranes coated by ink-jet printing of Pt/MWCNTs electrocatalyst for PEMFCs[J].International Journal of Hydrogen Energy,2012,37(15):11290-11298.
[29]BonakdarpourA,Stevens K,Vernstrom G D,et al.Oxygen reduction activity of Pt and Pt-Mn-Co electrocatalysts sputtered on nanostructured thin fi lm support[J].Electrochimica Acta,2007,53(2):688-694.
[30]Ahluwalia R K,Wang X,Lajunen A,et al.Kinetics of oxygen reduction reaction on nanostructured thin fi lm platinum alloy catalyst[J].Journal of Power Sources,2012,215:77-88.
[31]Sinha P K,Gu W,Kongkanand A,et al.Performance of nano structured thin film(NSTF)electrodes under partially-humidified conditions[J].Journal of The Electrochemical Society,2011,158(7):B831-B840.
[32]Vliet D,Wang C,Debe M,et al.Platinum-alloy nanostructured thin fi lm catalysts for the oxygen reduction reaction[J].Electrochimica Acta,2011,56(24):8695-8699.
[33]Advanced MEA’s for Enhanced Operating Conditions,Amenable to High Volume Manufacture[OL].3M/DOE Cooperative Agreement,No.DE-FC04-02AL67621,2003 DOE Hydrogen,fuel cells,and infrastructure technologies program review meeting,Berkeley,2003.http://www1.eere.energy.gov/hydrogenandfuelcells/pdfs/merit03/72_3m_mark_debe.pdf.
[34]Debe M K,Schmoeckel A K,Vernstrom G D,et al.High voltage stability of nanostructured thin fi lm catalysts for PEM fuel cells[J].Journal of Power Sources,2006,161(2):1002-1011.
[35]Sharma S,Pollet B G.Support materials for PEMFC and DMFC electrocatalysts:A review[J].Journal of Power Sources,2012,208:96-119.
[36]Wu G,Chen Y S,Xu B Q.Remarkable support effect of SWNTs in Pt catalyst for methanol electro oxidation[J].Electrochemistry Communications,2005,7(12):1237-1243.
[37]Gharibi H,Javaheri M,Mirzaie R A.The synergy between multi-wall carbon nanotubes and Vulcan XC-72R in microporous layers[J].International Journal of Hydrogen Energy,2010,35(17):9241-9251.
[38]Chang W C,Nguyen M T.Investigations of a platinumruthenium/carbon nanotube catalyst formed by a two-step spontaneous deposition method[J].Journal of Power Sources,2011,196(14):5811-5816.
[39]Rajalakshmi N,Ryu H,Shaijumon M M,et al.Performance of polymer electrolyte membrane fuel cells with carbon nanotubes as oxygen reduction catalyst support material[J].Journal of Power Sources,2005,140(2):250-257.
[40]Wu X X,Xu H F,Lu L,et al.The study on dynamic response performance of PEMFC with RuO2·xH2O/CNTs and Pt/C composite electrode[J].International Journal of Hydrogen Energy,2010,35(5):2127-2133.
[41]Carmo M,Paganin VA,Rosolen J M,et al.Alternative supports for the preparation of catalysts for low temperature fuel cells:The use of carbon nanotubes[J].Journal of Power Sources,2005,142(1-2):169-176.
[42]Zhang J W,Jiang D Z.Influence of geometries of multi-walled carbon nanotubes on the pore structures of Buckypaper[J].Composites Part A:Applied Science and Manufacturing,2012,43(3):469-474.
[43]Huang H,Zhang W K,Li M C,et al.Carbon nanotubes as a secondary support of a catalyst layer in a gas diffusion electrode for metal air batteries[J].Journal of Colloid and Interface Science,2005,284(2):593-599.
[44]Hu C G,Wang W L,Wang S X,et al.Investigation on electrochemical properties of carbon nanotubes[J].Diamond and Related Materials,2003,12(8):1295-1299.
[45]Waje M M,Wang X,Li W Z,et al.Deposition of platinum nanoparticles on organic functionalized carbon nanotubes grown in situ on carbon paper for fuel cells[J].Nanotechnology,2005,16(7):S395-S400.
[46]Zhu W,Ku D,Zheng J P,et al.Buckypaper-based catalytic electrodes for improving platinum utilization and PEMFC’s performance[J].Electrochimica Acta,2010,55(7):2555-2560.
[47]Zhu W,Zheng J P,Liang R,et al.Ultra-low Pt loading buckypaper electrocatalyst synthesized in supercritical fluid for PEMFC[C]//Electrochemical Society.2010 ECS-The Electrochemical Society,218th ECS Meeting,Meet.Abstr.MA2010-02 643.
[48]Kim K W,Kim S M,Choi S,et al.Electroless Pt deposition on Mn3O4nanoparticlesviathegalvanicreplacementprocess:Electrocatalytic nanocomposite with enhanced performance for oxygen reduction reaction[J].ACS Nano,2012,6(6):5122-5129.
