全氟甲基乙烯基醚合成的研究进展
2013-10-11贾文志朱志荣
贾文志,朱志荣
(同济大学化学系,上海 200092)
全氟甲基乙烯基醚(CF3OCF=CF2,简称PMVE)是合成氟橡胶和氟塑料的重要单体,由于其结构的特殊性,在开发新型综合性能优异的含氟聚合物中有着广泛的用途[1],特别是应用于目前需求量巨大的聚四氟乙烯中。聚四氟乙烯(PTFE)作为目前氟塑料的主要产品,广泛应用于航空航天、石油化工、机械、电子电器、建筑、纺织等诸多领域[2-3]。尽管PTFE具有良好的性能,但也存在一些缺陷,如力学性能较差、线膨胀系数较大、耐蠕变性差、耐磨性差、成型和二次加工困难等[4],使其应用范围受到一定的限制。将全氟甲基乙烯基醚少量地引入PTFE之后,可以改善其加工性能。全氟甲基乙烯基醚与PTFE按一定的比例共聚时,能形成新的聚合物材料,并且克服了PTFE自身的一些缺陷。因此,全氟甲基乙烯基醚的出现拓展了聚四氟乙烯的应用范围。
全氟甲基乙烯基醚是聚四氟乙烯重要的化学改性剂之一[5]。改性的 PTFE材料既保持了原 PTFE所具有物理力学性能和耐腐蚀性、耐候性等特性[6],还具有可熔融、抗冷流和耐折性、光泽性好等优点,加工使用非常方便,它是目前耐热性最好的氟橡胶。改性的PTFE材料具有很低的透气性,耐蠕变,适合作为强化学腐蚀环境下使用的密封件、膜片、软管等。全氟甲基乙烯基醚还可以与四氟乙烯和偏氟乙烯共聚,合成低温性能优良、可用过氧化物交联的偏氟醚橡胶[7]。除此之外,全氟甲基乙烯基醚还可以作为合成新型农药双苯氟脲的重要原料[8]。
全氟甲基乙烯基醚合成方法的研究始于 20世纪50年代,并主要集中在美国、德国、日本、俄罗斯等发达国家,我国对于全氟甲基乙烯基醚的合成研究尚处于起步阶段。目前关于合成全氟甲基乙烯基醚的技术文献报道很少,主要是以专利的形式公开,仅近来才有部分研究论文发表。本文归纳了近年来关于全氟甲基乙烯基醚的各种合成方法,分析了各种方法在工业应用上的优点和不足。通过对文献和专利进行总结和归类,将目前合成全氟甲基乙烯基醚的方法归纳为:①四氟乙烯法;②热分解法;③还原法;④六氟环氧丙烷法。下文针对上述4种合成方法进行详细介绍,并且分析其工业应用的利与弊。
1 四氟乙烯法
四氟乙烯法是指以四氟乙烯和三氟甲氧盐(CF3OM)进行取代反应生成全氟甲基乙烯基醚(PMVE)。该方法的理论依据最早在1959年美国专利 US2917548[1]中提出来,该专利采用四氟乙烯(CF2=CF2)和醇钠(RONa)为原料合成三氟乙烯基醚系列衍生物(ROCF=CF2),该方法反应通式:CF2=CF2+RONa —→ ROCF=CF2+ NaF。其中,该专利提到的R基团为甲基(—CH3)、1,1,1-三氟乙基(CF3CH2—)、1-正戊烯基(CH2=CHCH2CH2CH2—)、正丙基(CH3CH2CH2—)。该专利可以作为合成PMVE的一个设想,然而专利中提到的R基团没有包括—CF3,也没有提到合成PMVE的方法。由于CF3OM性质极不稳定,当时没有找到合适的合成途径,所以在该专利中无法将四氟乙烯法引入到PMVE的合成中。
近些年,科研工作者就三氟甲醇盐的合成展开研究,并且取得了突破性的进展,为四氟乙烯法合成PMVE起到推进的作用。周晓猛等[9]采用碳酰氟(COF2)与氟化盐在一定的条件下成功地合成了三氟甲醇盐。之后就用三氟甲醇盐(CF3OM)作为原料,取代四氟乙烯中的一个氟原子形成PMVE,该方法对上述美国专利中提到的反应通式进行了很好的补充。具体的反应过程如式(1)、式(2)。
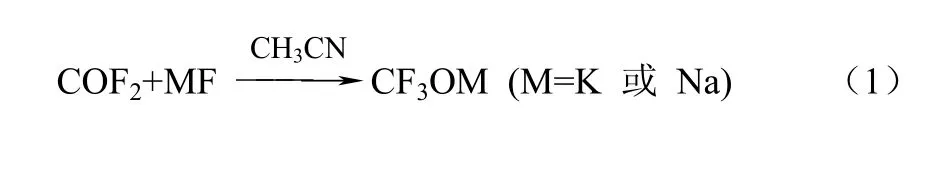
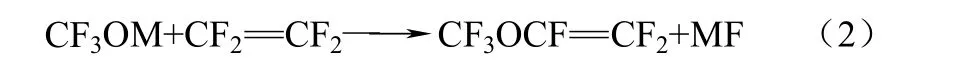
该合成方法分为两步:第一步是以碱金属氟化物(MF,M为K或Na)和碳酰氟在乙腈作为溶剂下,并常温、隔绝空气下制备三氟甲氧基盐;第二步在反应中存在少量水、常温下,四氟乙烯与三氟甲氧基盐在密闭的容器里反应72 h,通过色谱对收集的产物进行分析,PMVE的含量为72.5%。
该方法的优点是无需催化剂、常压下进行、反应条件温和,设备简单,生产成本较低。但是该方法工艺过程复杂,COF2难以回收利用,第一步的反应生成三氟甲氧基盐的产率低,第二步反应需要在一个无氧的环境下进行(四氟乙烯遇氧气有爆炸的危险),反应的时间过长。且该步反应需要加入少量水,而形成的游离态的金属氟化盐对反应釜有腐蚀性。
2 热解法
热解法是以全氟-2-甲氧基丙酰氟为原料,通过催化剂和碱金属盐两种方法来合成PMVE。对于全氟-2-甲氧基丙酰氟热解反应需要一个较高的温度,高活性的催化剂可以降低热解的温度,反应工艺简单、操作易于控制,而且催化剂可以多次反复使用。通过碱金属盐来热解全氟-2-甲氧基丙酰氟需要两步反应完成,而且碱金属盐无法重复利用。催化热解全氟-2-甲氧基丙酰氟法具有一定研究意义和很多可开发的空间,在工业生产上具有一定的优势,但是高效催化剂的研发是该方法中的技术难点。下文介绍不同催化剂的活性、碱金属盐脱酰氟能力以及脱羧酸盐的机理。
2.1 全氟-2-甲氧基丙酰氟催化热解
1967年,美国专利US3321532[10]报道了全氟-2-甲氧基丙酰氟(CF3OCF(CF3)COF,沸点10~12 ℃)催化热解生成PMVE。专利中以Ⅱ-A、Ⅱ-B、Ⅲ-A和Ⅳ-A族元素的氧化物作为催化剂,优先采用的催化剂是氧化锌、氧化钙、氧化铝、氧化钡、氧化铅和氧化硅,该类催化剂可以用于全氟烷基丙酰氟醚的催化热解。以 N2气稀释的全氟-2-甲氧基丙酰氟在催化剂床层中热解生成PMVE,其化学反应式如式(3)。

表1列出各种催化剂在不同反应条件下全氟-2-甲氧基丙酰氟的转化率和产物的产率情况。从表1中可以看出,氧化锌和氧化钙具有较好的催化活性,以氧化硅作为催化剂时,反应温度较高,且催化剂损失严重,因为部分形成CO2和SiF4而流失。当以氧化钙作为催化剂时,将接触时间0.5 min提高至4 min之后,产物中出现了全氟甲基乙基醚(CF3OCF2CF3),且全氟-2-甲氧基丙酰氟的转化率也降低至70%。同样以氧化钡为催化剂时,接触时间为3.7 min,原料的转化率为62%,PMVE的选择性为80%,CF3OCF2CF3的选择性为12%。
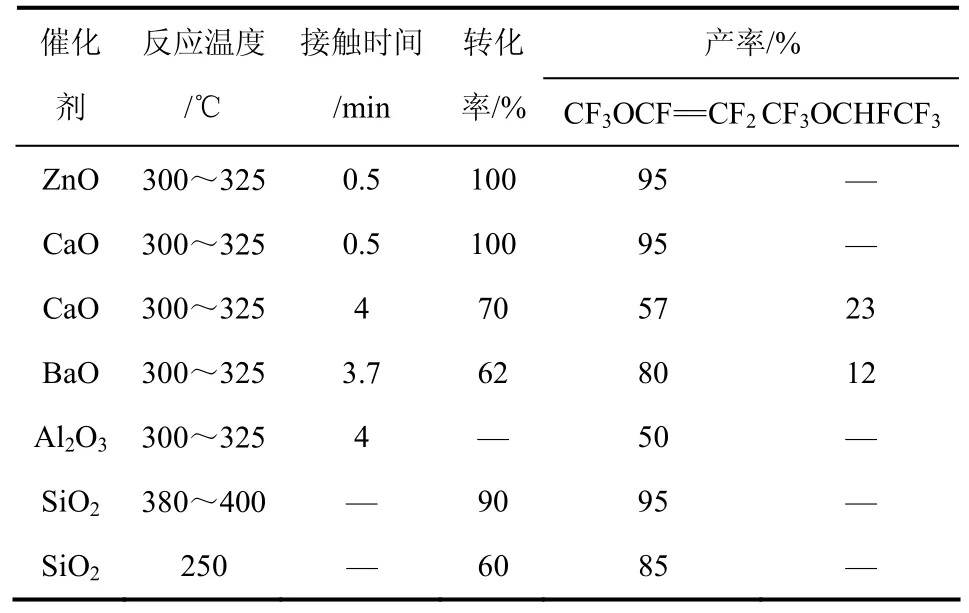
表1 不同催化剂对全氟-2-甲氧基丙酰氟热解反应的影响
当延长反应接触时间时,易产生三氟甲氧基四氟乙烷基醚副产物,这种副产物用普通的精馏方法很难分离,可能会使后续处理工艺的成本大幅提高。由上可见,该方法简单、易于控制,催化剂可回收利用,后期可以通过适当降低转化率、提高PMVE的选择性来减少副产物的生成。
2.2 全氟甲氧基丙酸盐热解
Lebedev等[11]报道了一种PMVE的合成方法,该方法是:在反应温度为120 ℃时,以二甘醇二甲醚作为溶剂,全氟-2-甲氧基丙酰氟和碳酸钠反应生成 CF3OCF2CF2COONa,然后CF3OCF2CF2COONa受热脱去酰氟基形成PMVE。当全氟-2-甲氧基丙酰氟与碳酸钠的摩尔比为 0.5时,最终产物中没有PMVE的生成,其中CF3OCF(CF3)COCF(CF3)OCF3占45%,CF3OCF(CF3)CF=CFOCF3占55%;当全氟-2-甲氧基丙酰氟与碳酸钠的摩尔比大于1时,主要产物为 PMVE(95%),CF3OCF(CF3)CF=CFOCF3为5%。该方法的化学反应式如式(4)。
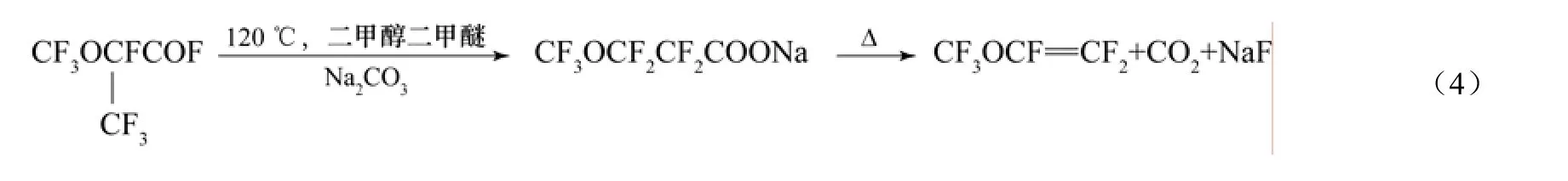
CF3OCF2CF2COONa在120 ℃时热解,通过脱羰基作用形成CF3OCF2CF2−和Na+,消去CF3OCF2CF2−中β位上的F−形成COF2和四氟乙烯,产物PMVE和四氟乙烯的产率分别为30%(摩尔分数)和60%(摩尔分数)。该文中比较了碳酸钠和碳酸钾对反应的影响,结果发现,当将K+取代Na+后,四氟乙烯的产率增加至 95%(摩尔分数),这说明了 CF3O−与 K+结合比较稳定,这与文献[12-13]的观点是一致的。
Gubanov等[12]将CF3OCF2CF2COONa在270 ℃高温热解时,反应产物中主要有 PMVE、四氟乙烯[14]和CF3OCF2CF2COF存在,各主要产物的摩尔比分别为 26%、24%和 35%。CF3OCF2CF2COONa高温热解的反应机理示意如式(5)。
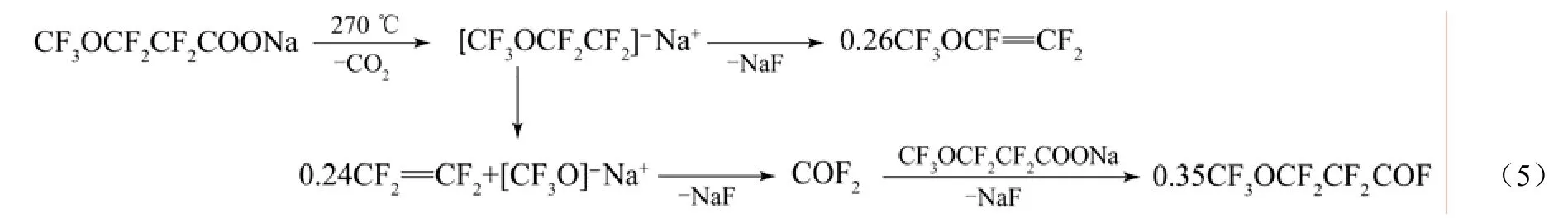
专利 US 3291843[15]、CN 101659602[16]中报道CnF2n-1OCF(CF3)COF系列衍生物在Ⅰ-A和Ⅱ-A族元素的碱金属盐中高温分解生成 CnF2n-1OCF=CF2。Fritz等[15]考察全氟-2-甲氧基丙酰氟及其衍生物在不同的碱金属盐下脱酰氟的情况,见表 2。在300 ℃下,稀释的全氟-2-甲氧基丙酰氟气体通过K2SO4和Na2SiO3床层,停留时间为10 min,热分解后生成 PMVE的产率为 60%和 95%。CnF2n-1OCF(CF3)COF系列衍生物在碱金属碳酸盐中的热分解机理与上述报道[12]类似,先形成羧酸盐(CnF2n-1OCF2CF2COOM),然后分解为 CnF2n-1OCF=CF2、MF和CO2;在碱金属硫酸盐中先形成CnF2n-1OCF2CF2COOM、MF和 SO3,最后CnF2n-1OCF2CF2COOM分解为目标产物。
专利CN 101659602[16]采用RfOCF(CF3)COF与碱金属盐/氢氧化物[摩尔比 1∶(2.05~2.2)]在无水或有机溶剂中反应生成含氟的羧酸盐,并经干燥除去水。然后在反应温度190~220 ℃、氮气情况下,含氟的羧酸盐热解1 h之后得到RfOCF=CF2。该专利中涉及以全氟-2-甲氧基丙酰氟原料合成 PMVE的具体实例,在不锈钢反应釜中加入20%(质量分数)的KOH水溶液,在搅拌下缓慢滴入全氟-2-甲氧基丙酰氟,反应温度为−10 ℃,反应30 min之后,将反应产物真空烘干,然后在 220 ℃下热解得到PMVE,产率为61.2%。
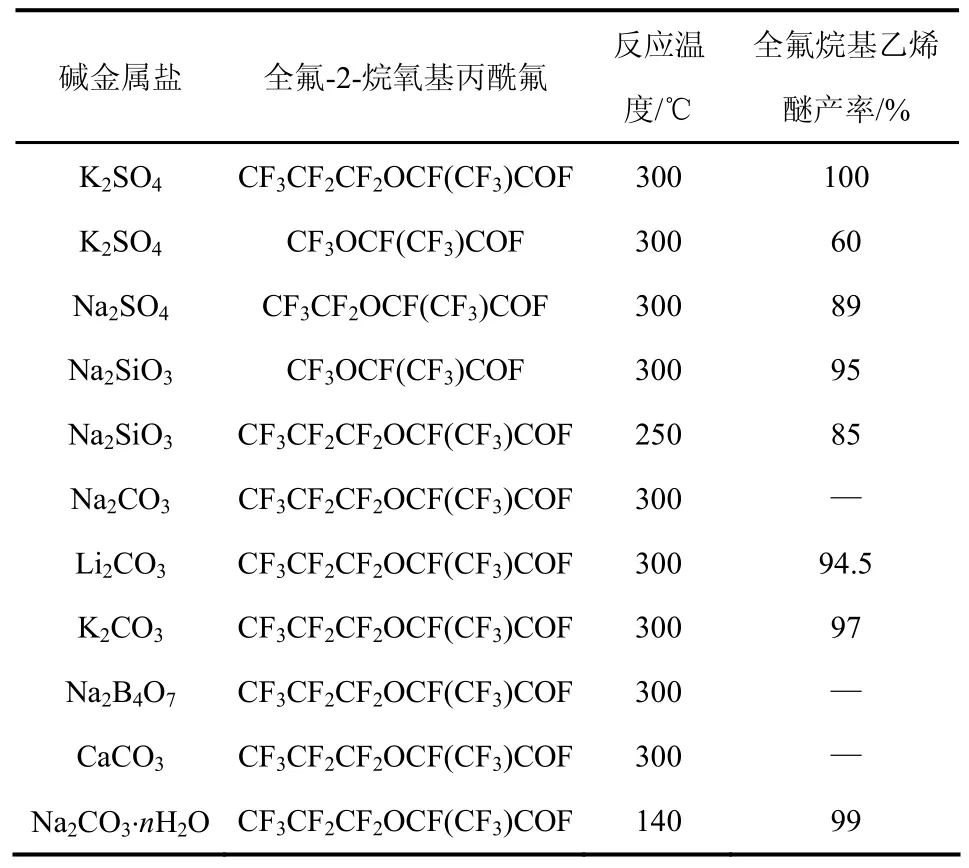
表2 全氟-2-烷氧基丙酰氟在不同碱金属盐作用下脱酰氟的结果
该方法无需催化剂,方法简单,但是该方法会消耗大量的碱金属碳酸盐、硫酸盐、氢氧化物等,获得PMVE产品的周期较长,并且生成的碱金属氟化物可能对环境产生污染。
3 还原法
3.1 金属锌还原法
金属锌能与含卤素的烷烃[17]、硅烷[18]以及羰基[19]发生反应,但是关于锌还原卤代氟醚(CF3OCX2CY3,X和 Y为卤素)生成烯烃氟醚(CF3OCX=CY2)的报道非常少,William 等[20]首次报道了锌粉还原三氟甲基-1,2-二氯-1,2,2-三氟乙基醚(CF3OCClFCClF2)生成PMVE,采用三氟甲基次氟酸(CF3OF)和 1,2-二氟-1,2-二氯乙烯(CFCl=CFCl)作为原料,通过两步合成目标产物。为了避免副反应的产生,第一步反应须在比较温和的条件下进行,而在较高的温度下有碳酰氟、二氧化碳和1,1,2,2-四氟-1,2-二氯乙烯(CF2ClCF2Cl)的生成。第二步反应在二甲亚砜溶剂(DMSO)的存在下,加入锌粉脱去HCl最后得到PMVE,产率为59.3%。该过程具体的反应式如式(6)、式(7)。
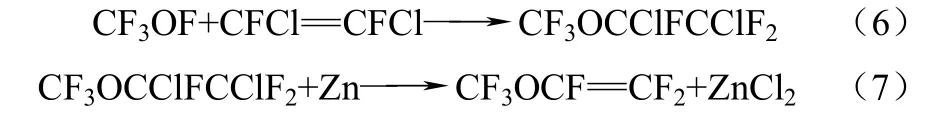
杨晓勇等[21]在William等合成PMVE方法的基础上进行改进,改进的手段是:在第二步反应锌粉还原三氟甲基-1,2-二氯-1,2,2-三氟乙基醚(CF3OCClFCClF2)时,选用 DMF作为溶剂能提高PMVE的产率。在对溶剂二甲基甲酰胺进行脱水处理和活化锌粉之后,可将产率提高到60%~80%。
碳酰氟需要通过一氧化碳与氟气两步氟化得到,由于该原料属于易燃、易爆物,且有剧毒,是一种危险的氟化试剂,从而限制了该原料的工业应用。此外,该方法的第二步采用锌粉还原,消耗大量的锌粉和溶剂,成本较高,并且锌粉对三氟甲基-1,2-二氯-1,2,2-三氟乙基醚的还原性能不高。
3.2 加氢脱氯法
氟氯烃(CFCs)和氢氟氯烃(HCFCs)是目前公认的大气臭氧消耗物质和温室效应气体[22-23]。加氢脱氯法是氟氯烃(CFCs)和氢氟氯烃(HCFCs)脱氯的重要方法之一[24-25]。加氢脱氯法无论是在工业应用上,还是作为基础研究,一直受人们的关注和重视,也是很多科研工作者研究的热点。近些年来,利用加氢脱氯法合成PMVE也开始引起了人们的研究兴趣。
2009年,Stefano等[26-27]报道了采用气相加氢法脱去 CF3OCFClCF2Cl中的 Cl,生成目标产物PMVE。该方法是以活性炭负载贵金属作为加氢催化剂,其中贵金属为镍和钌。关于Ni/C和Ru/C催化剂用于含氟氯烷烃和氟氯烯烃加氢反应的文献[28-31]已有报道,但对于CF3OCFClCF2Cl加氢的报道还是首次。Stefano等以1%(质量分数)Ru/C和1.5%(质量分数)Ni/C作为催化剂,反应20 h后的转化率分别为 23.4%和 14.9%,对应的选择性分别为97.4%和96.5%。反应式如式(8)。
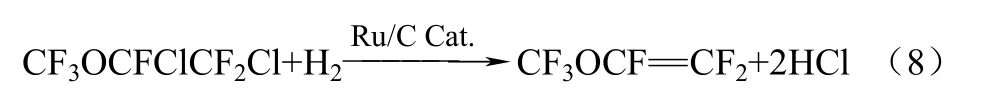
该方法操作简单,易于控制,对环境污染小,三废处理容易,反应原料可以经分离后循环利用,并且该反应可操控性好,但是该方法的最大弊端是原料不易获得,而且催化剂的转化率不高。
4 六氟环氧丙烷法
以六氟环氧丙烷和碳酰氟为原料可以制备PMVE。采用COF2与六氟环氧丙烷(沸点−42 ℃)反应得到全氟-2-甲氧基丙酰氟,然后全氟-2-甲氧基丙酰氟与K2CO3或Na2CO3形成羧酸盐,羧酸盐热解之后即可得到含氟乙烯基醚。具体的反应式如式(9)、式(10)。
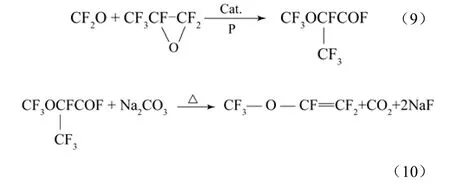
该方法的文献主要是以专利形式出现的,研究性的报道较少。专利US3114778[32]选用碱金属氟化物(如 CsF等)、活性炭、氟化银和氟化铵作为催化剂,以二甘醇二甲醚作为溶剂,反应温度为100~600 ℃,六氟环氧丙烷(HFPO)与CnF2n+1COF(全氟烷烃酰氟,n=0,1,2,··)在反应釜中反应形成全氟-2-甲氧基酰氟。然后在反应温度为 110~250 ℃下,将全氟-2-甲氧基酰氟通过Ⅰ-A族的金属碳酸盐或硫酸盐(主要有 K2CO3、Na2CO3、Li2CO3和Na2SO4等)脱去酰氟基团形成全氟烷烃乙烯基醚。
该专利是以碳酰氟和六氟环氧丙烷作为原料合成PMVE。其中第一步反应以活性碳为催化剂时,生成全氟-2-甲氧基丙酰氟的产率为33%,六氟环氧丙烷的转化率为 96.4%;第二步热解反应是在 300℃下以K2SO4为催化剂的固定床反应器上进行,得到PMVE的产率为60%。当第一步反应以氟化铯为催化剂、以二乙二醇二甲醚为溶剂时,在 75 ℃下反应4 h之后得到产率为68%的全氟-2-甲氧基丙酰氟;第二步将全氟-2-甲氧基丙酰氟与 KOH溶液作用形成2-甲氧基丙酸钾(CF3OCF2CF2COOK),再在200 ℃下加热24 h,可全部转化为PMVE目标产物。
该方法原料易得,PMVE的产率高,比较容易实现工业化生产。但是该方法还存在不足之处,第一步反应中全氟-2-甲氧基丙酰氟的选择性不高,副产物较多,第二步反应与上述的全氟甲氧基丙酸盐热解法是一样的,同样会消耗大量的碱金属碳酸盐、硫酸盐、氢氧化物等,并且生成的碱金属氟化物,腐蚀性强。
5 展 望
目前的几种合成PMVE的方法各有利弊,在工艺技术、催化剂、环保、原料来源等方面还存在一些问题,这也是制约现代氟化工发展的重要问题。对于目前合成 PMVE工业化问题的解决有两种途径可供选择:一是在现有的方法上进行创新、改进和完善;二是弃除旧的技术路线,开发新的合成工艺方法。
对于现有的技术方法改进创新,近些年来主要以专利的形式展现,并已有所进展,但是还有很多研究开发的空间。目前生产PMVE较有前景的方法是六氟环氧丙烷法,就目前现有的技术而言该方法相对较容易进行工业化生产,但是六氟环氧丙烷法还存在产物选择性不高,消耗大量的碱金属碳酸盐、硫酸盐、氢氧化物等,并且产生的腐蚀性强等问题。将六氟环氧丙烷法和全氟-2-甲氧基丙酰氟催化热解法的优点相结合,开发出高性能的催化剂,可以得到高产率和选择性、经济性高的PMVE,这将是未来PMVE工业化生产发展的重要方向。
目前关于 PMVE的合成方法的专利和文献报道还较少,近些年还未见一种全新的合成方法报道。由于PMVE含有一个—CF3(三氟甲基),研究合成PMVE的方法可以为全氟烷基乙烯基醚系列衍生物(CnF2n+1OCF=CF2)的合成技术路线提供借鉴。PMVE在开发新型综合性能优异的含氟聚合物中有着广泛的用途,特别是应用于目前PTFE工业生产中。那么,提高PMVE产率、产物易于分离提纯、降低能耗、减少副产物含量、降低设备强腐蚀和环境污染等问题将是未来PMVE工业化生产的难点。开发出 PMVE生产的新工艺技术不但能促进我国高性能PTFE材料开发与可持续发展,而且对我国不断发展中的氟橡胶产业的成长也具有重大的意义。
[1]Stanley D,Wilmington D.Chemical compouds and process for their preparation:US,2917548[P].1959-11-15.
[2]Scheirs J.Modern Fluoropolymers:High Performance Polymers for Diverse Applications[M].New York:John Wiley & Sons,1997.
[3]Ameduri B,Boutevin B.Well-architectured Fluoropolymers:Synthesis,Properties and Applications[M].Elsevier,2004.
[4]Ameduri B,Boutevin B.Update on fluoroelastomers:From perfluoroelastomers to fluorosilicones and fluorophosphazenes[J].Journal of Fluorine Chemistry,2005,126:221-229.
[5]袁海根,曾金芳,杨杰,等.聚四氟乙烯改性研究进展[J].塑料工业,2005,33(8):7-10.
[6]Blumm J,Lindemann A,Meyer M,et al.Characterization of PTFE using advanced thermal analysis techniques[J].International Journal of Thermophysics,2010,31(10):1919-1927.
[7]吴晓晋,缪建平,氟醚改性聚四氟乙烯的研制及其应用[J].化工生产与技术,2004,11(4):9-11.
[8]周彦峰,高中良,邱继平,等.杀虫剂双苯氟脲的合成研究[J].河南工业大学学报:自然科学版,2008(1):64-67.
[9]周晓猛,贾晓卿,鲍鹏,等.全氟甲基乙烯基醚的制备方法:中国,102211983[P].2011-04-08.
[10]Lorenz C E,Wilmington Del.Fluorocarbon ethers:US,3321532[P].1967-05-23.
[11]Lebedev N V,Berenblit V V,Starobin Y K,et al.Pyrolytic decarboxylation of some derivatives of perfluorinated mono-and dicarboxylic acids[J].Russian Journal of Applied Chemistry,2005,78(10):1640-1645.
[12]Gubanov V A,Zevakin I A,Gurari V É,et al.Influence of crown ethers on thermal decarboxylation of salts of perfluorocarboxylic acids[J].Zhurnal Organicheskoi Khimii,1981,17(8):1587-1590.
[13]Redwood M E,Willis C J.Fully fluorinated alkoxides.Part I.Trifluoromethoxides of alkali metals[J].Canadian Journal of Chemistry,1965,43(7):1893-1898.
[14]Ogden P H.Cyclisationsviafluoride ion induced isomerisations:A route to some novel perfluoroheterocyclic compounds[J].Journal of the Chemical Society C:Organic,1971,17:2920-2926.
[15]Fritz C G,Selman S.Fluorinated vinyl ethers and their preparation:US,3291843[P].1966-12-13.
[16]张鸣,孙凌云,杨旭仓,等.一种含氟乙烯基醚的制备方法:中国,101659602[P].2008-08-29.
[17]胡宏纹.有机化学[M].第3版.北京:高等教育出版社,2006:102-104.
[18]Seifert D A,Browning M F.Pilot-scale development of the zinc reduction process for production of high-purity silicon.Processing of energy and metallic[J].Processing of Energy and Metallic Minerals,1982,218(78):104-108.
[19]Gunther F A,Blinn R C.Dichlorodiphenyltrichloroethane.Ⅱ.Analogs.P,p’-DFDT and its degradation products[J].Journal of American Chemistry Society,1950,72(9):4282-4284.
[20]William S D,Eugene C S,Westmoreland G,et al.Polymers of fluorocarbon ethers and sulfides:Trifluoromethyl trifluorovinyl ether and sulfide[J].Journal of Polymer Science:Part A,1965(3):4065-4074.
[21]杨晓勇,周楠,黎爽,等.三氟甲基-1,1,2-三氟-1,2-二氯-乙基醚脱氯反应工艺:中国,1775722[P].2005-11-18.
[22]Jia W Z,Jin L Y,Wang Y J,et al.Fluorination of dichlorodifluoromethane to synthesize tetrafluoromethane over Cr2O3-AlF3catalyst[J].Journal of Industrial and Engineering Chemistry,2011,17(3):615-620.
[23]Makkee M,Sandt van de E J A X,Wiersma A,et al.Development of a satisfactory palladium on activated carbon catalyst for the selective hydrogenolysis of CCl2F2(CFC-12) into CH2F2(HFC-32)[J].Journal of Molecular Catalysis A:Chemical,1998,134(1-3):191-200.
[24]Moulijn J A,Makkee M,Wiersma A,et al.Selective hydrogenolysis of CCl2F2into CH2F2over palladium on activated carbon:Kinetic mechanism and process design[J].Catalysis Today,2000,59(3-4):221-230.
[25]Zheng X,Xiao Q,Zhang Y,et al.Deactivation of Pd/C catalysts in the hydrodechlorination of the chlorofluorocarbons CFC-115 and CFC-12[J].Catalysis Today,2011,175:615-618.
[26]Stefano M,Vito T,Giuseppe M.Method for manufacture perfluorovinylethers:WO,2009/150091[P].2009-12-17.
[27]斯特凡诺.米莱凡蒂,维托·陶特里,古赛佩·马奇奥尼.生产全氟乙烯基醚的方法:中国,102056877[P].2011-05-11.
[28]Wataru U,Satoshi T,Yutaka M,et al.Selective hydrodechlorination of CFC-113 to 1-chloro-1,2,2-trifluoroethylene over supported Ni catalysts[J].Chemistry Letters,1990(6):879-880.
[29]Mori T,Yasuoka T,Morikawa Y.Hydrodechlorination of 1,1,2-trichloro-1,2,2-trifluoroethane(CFC-113) over supported ruthenium and other noble metal catalysts[J].Catalysis Today,2004,88(3-4):111-120.
[30]Susumu O,Masatsune O,Hisashi H.Preparation of trifluoroethylene and chlorotrifluoroethylene:JP,3287552[P].1991-12-18.
[31]Yutaka M,Tsuneo I,Wataru U.Production of trifluorethylene:JP,5009138[P].1993-01-19.
[32]Fritz C G,Moore E P,Selman S,et al.Fluorinated vinyl ethers and their preparation:US,3114778[P].1963-12-17.
