并苯纳米环[6]CA及其衍生物的电子结构和光物理性质的密度泛函理论研究*
2013-09-27徐莹莹,阚玉和,武洁等
采用密度泛函理论PBE0方法在6-31G(d,p)基组水平上对比研究并六苯纳米环[6]CA及BN取代纳米环[6]CA-BN的几何结构及电子性质.同时探讨锂离子掺杂对不同体系的芳香性、前线分子轨道、电子吸收光谱及传输性质的影响.通过电离势、亲合势及重组能的计算,预测纳米环体系得失电子的能力及传输性能.结果表明:[6]CA的能隙很小,BN取代后,能隙明显增大;锂离子掺杂到两种纳米环中,在不明显改变前线分子轨道分布的前提下,几乎同步降低了最高占据轨道、最低未占据轨道能级,锂离子掺杂使载流子传输性能得到很大改善;电子吸收光谱拟合发现,BN取代使吸收光谱很大程度蓝移,吸收强度明显减小;而锂离子掺杂对光谱的强度及吸收范围没有明显影响.
1引言
自从1991年日本科学家Ijima[1]发现碳纳米管以来,纳米管的性质与结构的研究引起了人们极大的兴趣.碳纳米管在电学、光学和力学等方面具有奇异的特性,使其在纳米电子器件等方面具有广阔的应用前景[2,3].科学家关注的不仅仅是纳米家族独特的化学键和轨道,更重要的是如何通过改变环状结构尺寸的大小来改变它们潜在的化学和物理性质[4,5].因为它们是由具有放射状的p轨道的芳香性单元组成[5,6],可以沿着主链产生离域的电荷转移,被广泛作为电子传输材料应用在分子电子学和非线性光学(NLOs)等领域[7-9],使其成为近期的研究热点.单壁碳纳米管按手性分为扶手椅式(armchair)、锯齿式(zigzag)和螺旋式(chiral)三种[10,11].碳纳米管的导电性与其手性有密切关系,例如扶手椅式纳米管具有金属性,锯齿式纳米管具有半导体性[12].自1995年Zettl等[13]第一次发现BN纳米管结构后,科学家们采用多种方法成功制备了不同结构类型的BN纳米管[14-18].BN纳米管中B和N间的强离子键,使其不但有较高的化学稳定性,而且在电子结构上与碳纳米管存在显著的差别[19].对于半径较大的BN纳米管来说,无论其手性如何,都有接近5.5eV的禁带宽度[20,21],使它在高温、高强度纤维、半导体材料等方面有着更可能的实用性[22].正因为BN纳米管的特殊性能,所以对其展开研究是十分必要的.
碳纳米环作为碳纳米管的最小子单元[11,23],可体现纳米管的结构和性质.2008年,Jasti小组[24]首次合成了由苯基相连组成的环状体系,随后便引起对联苯纳米环的研究热潮,科学家致力于合成表征不同尺寸的联苯纳米环[25-30].根据最新文献报道,已成功合成的最小的联苯纳米环为[6]CPP[31].系列扶手椅式纳米环也陆续被合成[24,26,29,32,33].并苯纳米环作为锯齿式纳米管的最小单元,由于其非常小的能隙[6,34,35]以及结构的特殊性带来的比较大的环束缚能,使得并苯纳米环在合成上还没有得到突破.为了满足实际应用的需要,很多实验学家和理论学家致力于通过不同的方法改变和控制纳米环的电子性质.一方面,已有文献报道,BN取代形成的类并苯BN纳米环相比于并苯纳米环具有较大的能隙,使得BN纳米环具有更好的稳定性[19].另一个有效的途径是通过掺杂原子和离子来改变体系的电子性质[36-39].如锂离子嵌入碳纳米管中,可应用于锂离子电池负极材料中[40],其大的层间距使锂离子更容易嵌入和脱出,筒状结构在多次充放电循环后不会塌陷,这可以大大提高锂离子电池的性能和寿命[41].为了探讨CA,CA-BN纳米环及锂离子掺杂体系潜在的应用价值,本文选取较小的纳米环[6]CA和[6]CA-BN,采用密度泛函理论(DFT)对其基态几何结构、电子结构、吸收光谱、芳香性和载流子传输等多方面的性质进行讨论和分析,同时对比分析BN取代及锂离子掺杂对纳米环体系光物理性质的影响.
2 计算方法
采用杂化密度泛函PBE0方法,在6-31G(d,p)基组下对[6]CA,[6]CA-BN及锂离子掺杂体系进行全自由度基态几何优化,经过频率验证得到稳定构型.在此基础上,为了探讨锂离子在纳米环中不同位置的能量差异,采用PBE0/6-31G(d,p)方法,使锂离子在纳米环的轴向上以0.2A˚为步长做势能面扫描.芳香性讨论采用核独立化学位移(nucleus-independent chemical shift,NICS)判据[42],选取PBE0方法在6-31G(d,p)基组水平下计算每个稳定构型环中心及轴向上不同位点处的NICS值.应用含时密度泛函理论TD-PBE0/6-31G(d,p)方法研究纳米环体系激发态的电子性质,并拟合电子吸收光谱.为进一步指认光谱跃迁,了解吸收跃迁与结构的本质联系,我们对四个体系的自然跃迁轨道(NTO)[43]及跃迁密度矩阵(TDM)[44]进行讨论分析.
为探讨不同体系的电荷传输能力,采用PBE0/6-31G(d,p)和UPBE0/6-31G(d,p)方法分别优化得到中性分子和离子态分子的几何结构.在此基础上,通过单点计算,得到中性分子几何下的离子态和离子几何下中性态的能量,进而计算了垂直电离势VIP(VIP)及其电子亲合势VEA(VEA),绝热电离势AIP(AIP)及其电子亲合势AEA(AEA),空穴提取能PHE(HEP)和电子抽取能PEE(EEP).其中,垂直电离势是中性分子几何构型下阳离子和分子的能量差;绝热电离势是分别对中性分子和阳离子几何构型进行优化后的分子能量差;垂直电子亲合势是在中性分子几何构型下阴离子和分子的能量差;绝热电子亲合势是分别对中性分子和阴离子几何构型进行优化后的分子能量差;HEP是在阳离子几何构型下分子和阳离子的能量差;EEP是在阴离子几何构型下分子和阴离子的能量差.根据以下公式计算空穴重组能(λ+)和电子重组能(λ-)值.

上述计算均在Gaussian 09[45]程序包中完成.
3 结果与讨论
3.1 几何结构
采用PBE0泛函在6-31G(d,p)基组水平上对[6]CA,[6]CA-BN及锂离子掺杂体系进行几何优化,经频率验证后得到稳定的几何结构,如图1所示.部分几何参数列于表1.结果发现,[6]CA-BN和[6]CA相比,r1,r2和r3分别增加了约0.037,0.007和0.035A˚,使[6]CA-BN纳米环的B-N键长趋于平均化.r4增加了约0.107A˚,r5减小0.077A˚.[6]CA纳米环体系中,由于锂离子的掺杂,使r1,r2和r4略增长,而r3,r5键长略有减小.在[6]CA-BN纳米环中稍有不同,r1,r2和r5略微拉长,而r3和r4则略有减小.因此,BN取代相对锂离子掺杂对[6]CA纳米环的几何结构产生的影响更加明显.

表1 PBE0/6-31G(d,p)方法计算[6]CA,[6]CA-BN及锂离子掺杂体系的部分键长
从PBE0/6-31G(d,p)方法优化的结果可以看出,两种纳米环体系,尤其是[6]CA-BN纳米环,由于两个边缘化学环境完全不同,导致锂离子掺杂的最稳定位置并不在环中心.在Li+@[6]CA纳米环中,锂离子位于环中心上方约1.18˚A处,而在Li+@[6]CA-BN中,锂离子位于环中心上方0.46˚A处.为了探讨锂离子在纳米环中不同的掺杂位置带来的体系能量的差异,在全优化结构的基础上,锂离子在[6]CA及[6]CA-BN纳米环的Z轴方向上以0.2˚A为步长做势能面扫描,所得的能量曲线见图2.

图1 PBE0/6-31G(d,p)方法优化的[6]CA,[6]CA-BN及锂离子掺杂体系几何构型

图2 锂离子沿(a)[6]CA和(b)[6]CA-BN的Z轴穿过时的势能曲线
由图2可以发现,随着锂离子在纳米环轴向位置的不同,能量明显不同.由于未限制对称性,锂离子在穿过[6]CA纳米环时,形成一个不对称的势阱,锂离子位于Z轴方向上1.18A˚处达到势能最低点.对于[6]CA-BN纳米环,锂离子在轴向位置穿过时,形成三个不对称的势阱,分别在Z轴向上2.86,0.46,-1.34A˚处,位于0.46A˚处时能量最小,所以在0.46A˚时形成最稳定的结构.由势能曲线可以看出,锂离子在[6]CA-BN纳米环中任何一端穿过形成稳定的构型,都需要足够的能量去克服形成过程中的能量势垒,所以,Li+@[6]CA-BN纳米环一旦形成,将有很好的热稳定性.
3.2 芳香性
化合物的芳香性一直是理论领域研究的热点和难点之一,它对于有机化合物、无机化合物及团簇均有很好的适应性,NICS方法已经成为一种简便可行的测量NMR化学位移和研究分子芳香性的重要手段.
应用NICS判据,在全优化构型的基础上,采用GIAO-PBE0/6-31G(d,p)方法分析不同体系芳香性的大小.为了更清楚地分析不同位点处芳香性的变化,探讨了纳米环中心及轴向上不同位点处NICS值的大小.NICS计算位点和扫描方向如图3(a)所示.每个体系几何中心处的NICS(0)值列于表2.由表2可以看出,[6]CA纳米环的NICS(0)明显小于[6]CA-BN纳米环,所以其中心处的芳香性大于[6]CA-BN纳米环.同时,对比计算了锂离子掺杂体系,研究发现[6]CA纳米环由于锂离子掺杂,NICS(0)增加1.94 ppm(1 ppm=10-6),芳香性减小.相反,[6]CA-BN纳米环由于锂离子掺杂,NICS(0)明显减小(6.12 ppm),芳香性增大.计算Z轴方向上的NICS值表明(图3(b),3(c)),[6]CA纳米环的NICS值随位点距环中心距离的增加而增大,芳香性逐渐减小.而在[6]CA-BN纳米环中,NICS值随着距离的增加变化很小,数值接近零,所以芳香性很小.通过X轴方向上NICS值的变化表明(图3(d),3(e)),在[6]CA及[6]CA-BN纳米环中,随着位点距离环中心距离的增加,NICS值先减小后增大,分别在距离环中心1.0˚A和1.5˚A处,NICS值达到最小,数值为-37.23和-3.79 ppm.通过对比锂离子掺杂体系发现,锂离子掺杂使锂离子附近位点的NICS值明显减小,进而使芳香性显著增大.距离锂离子较远位点处的NICS值与掺杂前的NICS值相差不大,芳香性大小略微变化.

图3 PBE0/6-31G(d,p)计算的NICS值扫描示意图 (a)NICS值扫描方向示意;(b)[6]CA和Li+@[6]CA沿Z轴方向;(c)[6]CA-BN和Li+@[6]CA-BN沿Z轴方向;(d)[6]CA和Li+@[6]CA沿X轴方向;(e)CA-BN和Li+@[6]CA-BN沿X轴方向
3.3 前线分子轨道
前线分子轨道,尤其是最高占据轨道(HOMO)和最低未占据轨道(LUMO),作为电子结构性质研究的重要部分,对讨论激发态的性质,电子吸收光谱等具有重要作用.采用PBE0/6-31G(d,p)方法计算[6]CA,[6]CA-BN及锂离子掺杂体系的前线分子轨道能级.图4给出了四个纳米环的前线分子轨道能级和轨道分布,相关数据见表2.从前线分子轨道能级看出,[6]CA纳米环的能隙(ΔEH-L)较小(1.41 eV),电子由基态到激发态的跃迁更为容易.由于BN的取代,使[6]CA-BN的HOMO能级显著下降,LUMO能级显著上升,使得[6]CA-BN的能隙(6.01 eV)远远大于[6]CA的能隙,所以BN纳米管具有较高的化学稳定性.此外,通过对比发现,两种纳米环中引入锂离子,在不明显改变前线分子轨道电子密度分布的前提下,有效地改变了前线分子轨道能级,使HOMO,LUMO能级大幅度降低.另外,在Li+@[6]CA中,锂离子掺杂使能隙略增加了0.08 eV,相反,在Li+@[6]CA-BN中,锂离子的掺杂使能隙相对减小了0.71 eV.有趣的是,锂离子掺杂前后,前线轨道的电子密度分布和能隙变化很小,而HOMO,LUMO能级发生显著变化,另外,通过外加点电荷的计算,证实这主要是由于锂离子静电作用引起的.综上可知,BN取代使[6]CA纳米环的能隙明显增加,锂离子的掺杂显著降低了两种纳米环的前线分子轨道能级,而且削弱了[6]CA纳米环及[6]CA-BN纳米环的能隙差.
由于并苯纳米环的单态能很小,具有较高的反应活性,我们对比计算[6]CA,[6]CA-BN及锂离子掺杂体系的单态能,探讨BN取代及锂离子的掺杂对单态能的影响.相关数据见表2.通过计算表明,[6]CA的单态能(ΔES)相对很小,仅为0.95 eV.由于BN的引入,使[6]CA-BN的单态能(4.80 eV)明显增大.通过对比锂离子掺杂体系发现,锂离子的加入使Li+@[6]CA的单态能仅增大0.01 eV.相反,在Li+@[6]CA-BN中,锂离子的掺杂使单态能明显减小(0.82 eV).由此可知,[6]CA纳米环及锂离子掺杂体系的单态能较小,形成激发态较容易,所以基态不稳定.而[6]CA-BN纳米环及锂离子掺杂体系的单态能就相对较大,进而具有相对稳定的基态.这一结论与前线分子轨道分析得出的结论一致.
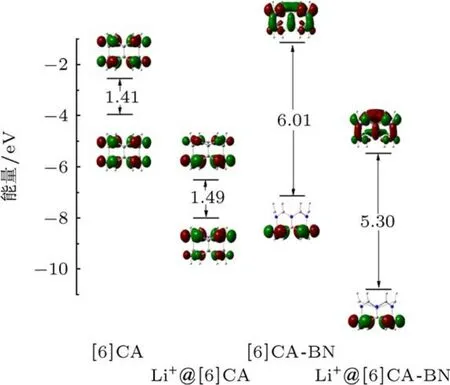
图4 [6]CA,[6]CA-BN及锂离子掺杂体系的前线轨道能级及电子密度分布

表2 四个纳米环体系的HOMO(E H),LUMO(E L),ΔE H-L,ΔE S(eV)和NICS(0)值
从前线分子轨道图(图4)可以看出,[6]CA纳米环的HOMO和LUMO比较离域,均匀地分布在两侧碳上,掺杂锂离子后,离域特征发生改变,HOMO的电子密度分布略偏向距离锂离子较近的碳原子上,而LUMO略偏向距离锂离子较远的碳原子上.由此可见,锂离子的掺杂,对[6]CA纳米环体系的前线分子轨道组成影响较小.对于[6]CA-BN纳米环体系,HOMO的电子密度主要定域在电负性比较大的N原子上,而LUMO电子密度分布相对离域,分布在整个纳米环体系上,锂离子的掺杂并没有明显改变其前线分子轨道的分布,因此由HOMO到LUMO跃迁形成的激发态具有电荷转移跃迁特征.
3.4 重组能
电离势与亲和势(EA)的大小可用来评估电子和空穴的注入能力,可以反映体系得失电子的难易程度.EA和EEP越大,越易接受电子,而电离能(IP)和HEP越小,越容易失去电子.由表3数据可以看出,在[6]CA,[6]CA-BN两种纳米环体系中,锂离子掺杂后,无论是垂直亲合势还是绝热亲合势都增大,所以锂离子的掺杂使体系得电子的能力增大,这与前面计算得到的前线分子轨道LUMO能级的降低相一致.从垂直电离能和绝热电离能来看,锂离子的掺杂使体系的电离能明显增大,给电子能力降低,这与HOMO能级降低趋势结果相符合.
重组能作为影响传输性能的一个重要因素,其值越小,载流子传输能力越强.从表3数据可以看出,[6]CA纳米环的电子重组能(0.25 eV)和空穴重组能(0.23 eV)很相近,所以[6]CA纳米环的电子和空穴的传输能力相当,可作为潜在的双极性传输材料,这与HOMO和LUMO电子密度的离域特征一致.对比发现,锂离子掺杂使电子重组能和空穴重组能都减小,所以锂离子的掺杂使体系的电子和空穴传输性能都有所改善.而在[6]CA-BN纳米环中,空穴重组能(0.54 eV)明显大于电子重组能(0.37 eV),因此空穴传输能力小于电子传输能力,这与HOMO分布定域,而LUMO分布离域相一致.而锂离子的掺杂使空穴重组能略微减小(0.03 eV),电子重组能却是明显增加(0.16 eV),所以,锂离子的掺杂使纳米环体系的空穴传输能力增加,电子传输能力降低.也就是说,锂离子的掺杂使Li+@[6]CA-BN体系趋向作为双极性传输材料.

表3 PBE0/6-31G(d,p)方法计算的亲合势,电离势,重组能,空穴提取能和电子抽取能
3.5 电子吸收光谱
为了探讨[6]CA,[6]CA-BN纳米环体系的光谱性质及锂离子掺杂对纳米环光谱强度的影响,我们计算了四个纳米环体系的电子吸收光谱,目前这些体系的光谱研究未见实验报道.尽管TDDFT是目前计算跃迁能的最广泛的方法,但是一般情况下,不同泛函的计算结果有较大的差异[46].所以,本文在PBE0/6-31G(d,p)全优化几何结构的基础上,运用多种密度泛函方法计算体系的电子跃迁光谱.分别采用GGA泛函BP86,杂化泛函B3LYP,PBE0,BH&HLYP,M06-2X及长程校正泛函CAM-B3LYP和LC-ωPBE多种密度泛函方法,在6-31G(d,p)基组水平上计算了[6]CA的电子吸收光谱,各泛函拟合所得吸收光谱如图5和表4.从图5可以看出,随着HF交换能成分的增加,不同泛函计算的吸收光谱出现不同程度的蓝移,吸收强度也明显不同.重要的是,通过对比发现不同泛函拟合的光谱图都有三个明显的吸收峰并且峰形基本一致.此外,根据已有文献报道,HF交换能含量适中的杂化泛函PBE0更适合共轭聚合物以及碳纳米管激发能的计算[47,48],因此在下面的计算中,我们选择杂化泛函PBE0方法对此类体系电子吸收光谱进行讨论分析.

表4 不同密度泛函方法计算的[6]CA的最大吸收波长及跃迁能
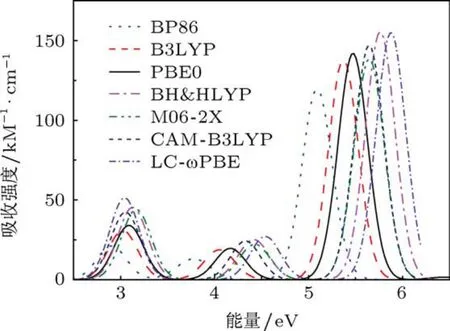
图5 体系[6]CA采用不同泛函在6-31G(d,p)基组水平计算拟合的电子吸收光谱

图6 PBE0/6-31G(d,p)方法计算拟合的电子吸收光谱
采用PBE0/6-31G(d,p)方法计算[6]CA,[6]CABN纳米环及锂离子掺杂体系的电子吸收光谱曲线,拟合的吸收光谱见图6.振子强度、吸收波长、激发能及主要轨道跃迁贡献见表5.从图6可以看出[6]CA纳米环及锂离子掺杂体系在紫外可见光范围内有三个明显的吸收峰,[6]CA纳米环的最大吸收峰位于226 nm处(5.49 eV),其主要轨道跃迁贡献由HOMO-3→LUMO+2(32%)和HOMO-1→LUMO+6(58%)组成.而[6]CA-BN纳米环与[6]CA纳米环的光谱明显不同,其原因是由于[6]CA-BN与[6]CA纳米环的能隙差别较大.[6]CA-BN纳米环的吸收光谱主要分布在远紫外区域,最大吸收峰位于169 nm处(7.34 eV),由HOMO-2→LUMO+5(81%)和HOMO→LUMO+4(10%)轨道跃迁贡献得来.锂离子的掺杂,对[6]CA纳米环的吸收光谱的影响很小,使[6]CA-BN吸收光谱略微红移,最大吸收峰位于183 nm处.
四个体系最大吸收峰处的主要跃迁轨道如表5所示,由于跃迁轨道贡献比较分散,对轨道的指认相对困难,为了更清楚地揭示吸收跃迁与结构的本质联系,我们对四个体系的NTO及最大吸收波长处TDM进行讨论分析,结果如图7所示.NTO分析结果表明,四个体系空穴到电子的跃迁都有明显的电荷转移,[6]CA纳米环最大吸收峰处的电子密度分布比较离域,主要跃迁被指认为π→π∗的电荷转移,锂离子的掺杂没有明显改变电子密度分布.而[6]CA-BN纳米环在最大吸收峰处的电荷转移比较复杂,主要跃迁指认为从电负性大的N原子到缺电子的B原子的电荷转移.锂离子的掺杂明显改变了BN纳米环的电子密度分布,使轨道分布更加定域.
尽管碳纳米环与BN纳米环有相似的结构,但是,两种纳米环体系中密度矩阵的离域模式却存在明显差别.从最大吸收波长处的电子TDM分析可以得出,[6]CA纳米环及锂离子掺杂体系,轨道分布相对离域,明显体现出苯单元内及苯单元间的电荷转移特性.而[6]CA-BN纳米环电子云分布明显定域,主要表现为硼氮环单元内的电荷转移.[6]CA-BN纳米环由于锂离子的掺杂,使电荷转移更加复杂,明显增加了硼氮环单元间的电荷转移.
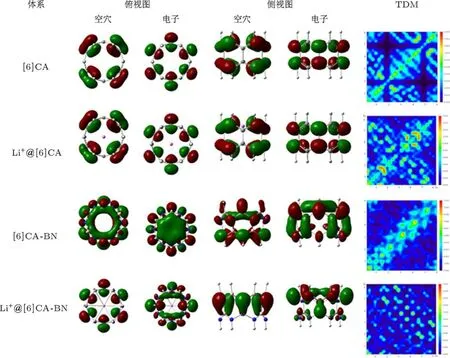
图7 [6]CA,[6]CA-BN及锂离子掺杂体系的NTO及TDM
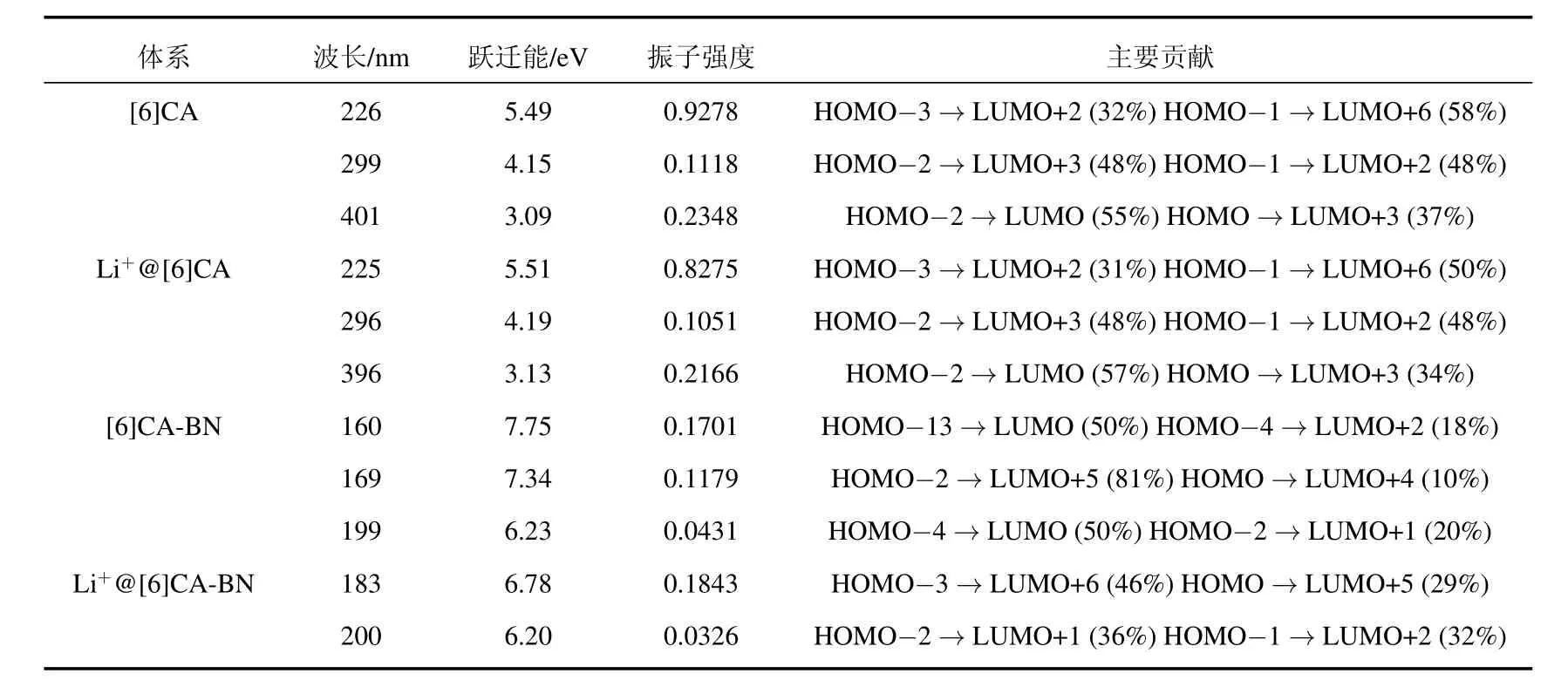
表5 PBE0/6-31G(d,p)方法计算四个体系的吸收波长,振子强度及主要跃迁形式
4 结论
为了探讨CA,CA-BN纳米环及锂离子掺杂体系潜在的应用价值,本文采用PBE0/6-31G(d,p)方法对比研究了[6]CA,[6]CA-BN纳米环及锂离子掺杂体系的几何结构、芳香性、电子吸收光谱及传输性能等光物理性质.研究结果表明:[6]CA纳米环的芳香性大于[6]CA-BN纳米环,这与[6]CA的轨道分布比较离域,[6]CA-BN的轨道分布较定域相符合.锂离子的掺杂明显减小了锂离子周围的NICS值,进而增大芳香性.前线分子轨道能级的计算表明,[6]CA的能隙较小,电子由基态到激发态的跃迁更为容易.BN取代使纳米环的HOMO下降、LUMO上升,所以[6]CA-BN的能隙(6.01 eV)远远大于[6]CA能隙.锂离子掺杂到这两种纳米环中,在不明显改变前线分子轨道分布的前提下,使HOMO,LUMO能级同时降低.锂离子掺杂使[6]CA的能隙略增加(0.08 eV),相反,使[6]CA-BN的能隙大幅度减小(0.71 eV),所以锂离子的加入削弱了[6]CA及[6]CA-BN的能隙差.重组能的计算表明,[6]CA可作为潜在的双极性传输材料,锂离子的掺杂使体系的电子和空穴传输性能都增加.[6]CA-BN的电子传输能力大于空穴传输能力,掺杂锂离子后,电子传输能力降低,空穴传输能力增加,使其倾向于双极性传输.通过拟合电子吸收光谱发现,[6]CA的主要吸收位于紫外可见光范围内,而[6]CA-BN的主要吸收位于远紫外区域.[6]CA中掺杂锂离子,对吸收光谱没有明显的影响.[6]CA-BN中掺杂锂离子,吸收光谱略微红移.跃迁密度矩阵分析表明,锂离子的掺杂增加了苯(硼氮)环单元间的电荷转移.综合以上计算可知,BN取代及锂离子的掺杂,有效地改变了纳米环的芳香性,电子性质及载流子传输性能.而锂离子掺杂对能隙的影响不大,导致锂离子的掺杂没有使吸收光谱发生明显变化.这些信息可为设计和制备新型的高效的纳米器件提供理论依据.
[1]Iijima S1991 Nature354 56
[2]Prasek J,Drbohlavova J,Chomoucka J,Hubalek J,Jasek O,Adam V,Kizek R 2011 J.Mater.Chem.21 15872
[3]Omachi H,Segawa Y,Itami K 2012 Acc.Chem.Res.45 1378
[4]Kawase T,Kurata H 2006 Chem.Rev.106 5250
[5]Scott L T 2003 Angew.Chem.Int.Ed.42 4133
[6]Eisenberg D,Shenhar R,Rabinovitz M 2010 Chem.Soc.Rev.39 2879
[7]Marsden JA,Miller JJ,Shirtcliff L D,Haley M M 2005 J.Am.Chem.Soc.127 2464
[8]Zhao T,Liu Z,Song Y,Xu W,Zhang D,Zhu D 2006 J.Org.Chem.71 7422
[9]Xu H L,Zhong R L,Yan L K,Su Z M 2012 J.Phys.Org.Chem.25 176
[10]Jasti R,Bertozzi CR 2010 Chem.Phys.Lett.494 1
[11]Bunz U H F,Menning S,Martin N 2012 Angew.Chem.Int.Ed.51 7094
[12]Dai HJ2002 Acc.Chem.Res.35 1035
[13]Chopra N G,Luyken RJ,Cherrey K,Crespi V H,Cohen M L,Louie SG,Zettl A 1995 Science269 966
[14]Golberg D,Bando Y,Eremets M,Takemura K,Kurashima K,Yusa H 1996 Appl.Phys.Lett.69 2045
[15]Han W Q,Bando Y,Kurashima K,Sato T 1998 Appl.Phys.Lett.73 3085
[16]Golberg D,Bando Y,Han W,Kurashima K,Sato T 1999 Chem.Phys.Lett.308 337
[17]Golberg D,Bando Y,Kurashima K,Sato T 2000 Chem.Phys.Lett.323 185
[18]Ma R,Bando Y,Sato T 2001 Chem.Phys.Lett.337 61
[19]Loh K P,Yang SW,Soon JM,Zhang H,Wu P 2003 J.Phys.Chem.A 107 5555
[20]Jia JF,Wu H S 2006 Acta Phys.Chim.Sin.22 1520(in Chinese)[贾建峰,武海顺2006物理化学学报22 1520]
[21]Sun WG,Liu X Y,Wang CY,Tang Y J,Wu WD,Zhang H Q,Liu M,Yuan L,Xu JJ2009 Acta Phys.Sin.58 1126(in Chinese)[孙卫国,刘秀英,王朝阳,唐永建,吴卫东,张厚琼,刘淼,袁磊,徐嘉靖2009物理学报58 1131]
[22]Jia JF,Wu H S,Jiao H J 2004 Acta Chim.Sin.62 1385(in Chinese)[贾建峰,武海顺,焦海军2004化学学报62 1385]
[23]Gleiter R,Esser B,Kornmayer SC 2009 Acc.Chem.Res.42 1108
[24]Jasti R,Bhattacharjee J,Neaton JB,Bertozzi CR 2008 J.Am.Chem.Soc.130 17646
[25]Steinberg B D,Scott L T 2009 Angew.Chem.Int.Ed.48 5400
[26]Takaba H,Omachi H,Yamamoto Y,Bouffard J,Itami K 2009 Angew.Chem.Int.Ed.48 6112
[27]Omachi H,Matsuura S,Segawa Y,Itami K 2010 Angew.Chem.Int.Ed.49 10202
[28]Yamago S,Watanabe Y,Iwamoto T 2010 Angew.Chem.Int.Ed.49 757
[29]Sisto T J,Golder M R,Hirst E S,Jasti R 2011 J.Am.Chem.Soc.133 15800
[30]Ishii Y,Nakanishi Y,Omachi H,Matsuura S,Matsui K,Shinohara H,Segawa Y,Itami K 2012 Chem.Sci.3 2340
[31]Xia J,Jasti R 2012 Angew.Chem.Int.Ed.51 2474
[32]Segawa Y,Miyamoto S,Omachi H,Matsuura S,ˇSenel P,Sasamori T,Tokitoh N,Itami K 2011 Angew.Chem.Int.Ed.50 3244
[33]Jasti R,Sisto T 2012 Synlett.23 483
[34]Chen Z,Jiang D E,Lu X,Bettinger H F,Dai S,Schleyer PV,Houk K N 2007 Org.Lett.9 5449
[35]Choi H S,Kim K S 1999 Angew.Chem.Int.Ed.38 2256
[36]Chen Y K,Liu L V,Tian WQ,Wang Y A 2011 J.Phys.Chem.C 115 9306
[37]Wang X,Liew K M 2012 J.Phys.Chem.C 116 1702
[38]Wang C,Wang L G,Zhang H Y,Terence K SW 2010 Acta Phys.Sin.59 536(in Chinese)[王畅,王利光,张鸿宇,Terence K SW 2010物理学报59 536]
[39]Yang C,Zhang B X,Feng Y F,Yu Y 2009 Acta Phys.Sin.58 4066(in Chinese)[杨春,张变霞,冯玉芳,余毅2009物理学报58 4066]
[40]Baibarac M,Lira Cant´u M,Or´o Sol´e J,Casa˜n Pastor N,Gomez Romero P 2006 Small 2 1075
[41]Liu Y Q 2010 Organic Nano and Molecular Devices(Beijing:Science Publishing House)p101(in Chinese)[刘云圻2010有机纳米与分子器件(北京:科学出版社)第101页]
[42]Schleyer PV,Maerker C,Dransfeld A,Jiao H J,Hommes N 1996 J.Am.Chem.Soc.118 6317
[43]Martin RL 2003 J.Chem.Phys.118 4775
[44]Tretiak S,Mukamel S2002 Chem.Rev.102 3171
[45]Frisch M J,Trucks G W,Schlegel H B,Scuseria G E,Robb M A,Cheeseman JR,Scalmani G,Barone V,Mennucci B,Petersson G A,Nakatsuji H,Caricato M,Li X,Hratchian H P,Izmaylov A F,Bloino J,Zheng G,Sonnenberg JL,Hada M,Ehara M,Toyota K,Fukuda R,Hasegawa J,Ishida M,Nakajima T,Honda Y,Kitao O,Nakai H,Vreven T,Montgomery Jr JA,Peralta JE,Ogliaro F,Bearpark M,Heyd JJ,Brothers E,Kudin K N,Staroverov V N,Kobayashi R,Normand J,Raghavachari K,Rendell A,Burant JC,Iyengar SS,Tomasi J,Cossi M,Rega N,Millam JM,Klene M,Knox JE,Cross JB,Bakken V,Adamo C,Jaramillo J,Gomperts R,Stratmann RE,Yazyev O,Austin A J,Cammi R,Pomelli C,Ochterski JW,Martin R L,Morokuma K,Zakrzewski V G,Voth G A,Salvador P,Dannenberg JJ,Dapprich S,Daniels A D,Farkas O,Foresman JB,Ortiz JV,Cioslowski J,Fox DJ 2009 Gaussian 09(Revision A.02 Gaussian,Inc.Wallingford CT)
[46]Zhao Y,Truhlar D G 2008 Acc.Chem.Res.41 157
[47]Scholes G D,Tretiak S,McDonald T J,Metzger W K,Engtrakul C,Rumbles G,Heben M J2007 J.Phys.Chem.C 111 11139
[48]Wong B M 2009 J.Phys.Chem.C 113 21921
