基于胆固醇的新型小分子胶凝剂的合成与胶凝行为
2013-09-17薛敏苗青房喻,*
薛 敏 苗 青 房 喻,*
(1西安文理学院化学与化学工程学院,西安710065;2陕西师范大学化学化工学院,应用表面与胶体化学教育部重点实验室,西安710062)
1 引言
凝胶是大量液体被少量溶质固定而形成的一类重要的软物质.尽管凝胶在高分子体系中很常见,但对小分子凝胶的研究要晚得多.1−5小分子凝胶是小分子胶凝剂(LMMGs)通过范德华相互作用、氢键、6π−π堆积1,7及静电作用8等分子间弱相互作用自组装形成一维结构,继而经过交联缠绕形成三维网络结构而使有机溶剂或水失去流动性而形成的.与高分子凝胶相比小分子凝胶具有很多优点,比如胶凝剂的分子结构确定,胶凝过程完全可逆等.利用这些性质可以设计众多功能凝胶体系并且可以制备更复杂的、结构可控的纳米材料.除了热可逆相变特性、微观和介观尺度的高分散性外,通过在小分子胶凝剂结构上引入相应的功能基团,小分子凝胶还有可能获得对光、电、pH值、氧化还原作用、主客体作用等的刺激响应性,因此在药物输运、传感和敏感器件制备、海上凝油、无机微纳米材料制备甚至“智能”推进剂等方面均表现出巨大的应用前景.
众所周知,由于胆固醇衍生物具有刚性骨架、多手性中心及强的范德华堆积等特点,因此一直是小分子胶凝剂研究的热点,得到了最为广泛的研究.1,9近十几年来,大量具有不同芳香基团和连接臂的胆固醇型小分子胶凝剂被合成和研究.虽然芳香基团并不是胶凝所必须的,但是它能够精细调节自组装超分子结构中分子的组织排列,从而调节凝胶材料的自组装性质.此外,π-结构的引入有可能伴随新的光电性质的出现,从而为新的功能材料创制奠定基础.
根据分子结构中胆固醇片段的个数,包含芳环结构的胆固醇类小分子胶凝剂被分成了两类:ALS和A(LS)2,其中A代表芳香基团,L代表连接臂,S代表胆甾基.相对于ALS结构,A(LS)2结构的出现为构建具有特殊性质的新型小分子胶凝剂提供了新的空间.10−13在已经报道的A(LS)2型小分子胶凝剂中,A多为具有大共轭体系的多环芳烃结构,以苯环等小芳香环为A部分的A(LS)2型小分子胶凝剂研究得很少.本小组在已有的以单个苯环为A结构的A(LS)2型小分子胶凝剂研究的基础上,14−16设计制备了连接臂包含丙二胺结构,具有不同取代位置的3种以苯环为A结构的A(LS)2型胆固醇类小分子胶凝剂,考察了这3种胶凝剂在常见溶剂中的胶凝行为,研究了取代基位置对此类化合物胶凝行为的影响.
2 实验部分
2.1 试剂与仪器
丙二胺、胆固醇氯甲酸酯(≥97%)和间苯二甲酰氯(≥98%)均购自阿法埃莎(天津)化学有限公司公司,未经纯化直接使用.邻苯二甲酸和对苯二甲酰氯均为上海国药集团产品(分析纯),二环己基羰二亚胺(DCC)和4−二甲氨基吡啶(DMAP)为分析纯.三乙胺用氢氧化钠除水后蒸馏使用;四氢呋喃经钠片回流后蒸馏;二氯甲烷、正己烷经无水CaCl2干燥后蒸馏,乙酸乙酯、甲醇和丙酮使用前经蒸馏纯化;实验用水均经离子交换,再二次蒸馏纯化.浓硫酸、盐酸等未经纯化直接使用.
Equinox 55型傅里叶变换红外光谱仪(德国Bruker公司),KBr压片;Avance 300型超导核磁共振波谱仪(瑞士Bruker公司),以四甲基硅烷(TMS)为内标;Vario EL III型元素分析仪(德国元素分析系统公司);Quanta 200环境扫描电镜(荷兰Philips-FEI公司);ALPHA1-2型冷冻干燥机(德国Christ公司);D/Max-3c型全自动X射线衍射仪(日本Rigaku公司).
2.2 实验过程
2.2.1 胶凝剂的合成与表征
3种胶凝剂的合成过程如图1所示,具体的合成步骤如下.
2.2.1.1 化合物a的合成
将3.3 mL(40 mmol)的丙二胺和0.29 mL(2 mmol)的三乙胺加入150 mL经纯化的二氯甲烷,然后在273 K搅拌所得到的混合物溶液.以50 mL二氯甲烷溶解0.9 g(2 mmol)胆固醇氯甲酸酯,再将此溶液缓慢滴加到上述丙二胺和三乙胺溶液中.滴加完后,将所得混合物在室温下搅拌10 h,过滤,滤液用水洗涤5次,再用无水硫酸镁干燥,旋转蒸发除去二氯甲烷.将所得到的固体用20 mL二氯甲烷溶解后,再向其中加入20 mL甲醇,产生少量白色絮状沉淀,将此沉淀过滤后,将滤液旋干,真空干燥后得到的白色固体即为化合物a,产率为60%.化合物a的表征结果:1H NMR(CDCl3/TMS,300 MHz):δ 5.36(s,1H,alkenyl),5.14(s,1H,COONH),4.48(m,1H,oxycyclohexyl),3.263(q,2H,NHCH2),2.77(t,2H,NH2CH2),0.69−2.33(m,47H,cholesteryl protons,NH2,CH2,).元素分析(C31H54N2O2),计算值(%):C,76.49,H,11.18,N,5.75.测定值(%):C,76.02;H,11.26;N,5.08.
2.2.1.2 化合物1的合成
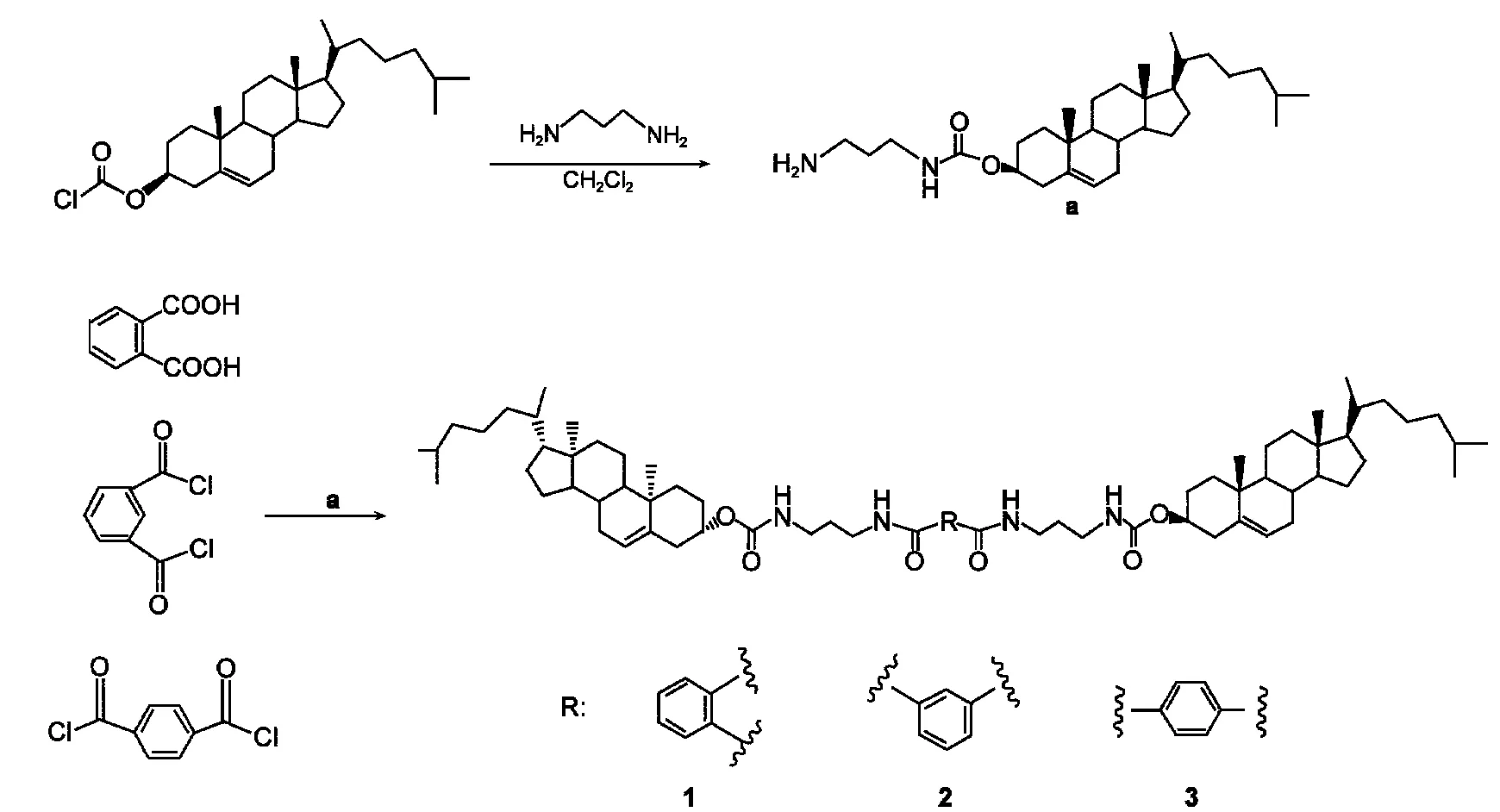
图1 化合物1−3的合成路线Fig.1 Synthesis routes of compounds 1−3
将0.498 g(3 mmol)的邻苯二甲酸和2.92 g(6 mmol)的化合物a用100 mL纯化后的四氢呋喃溶解后,在冰浴下搅拌.将1.24 g(6 mmol)的DCC和0.073 g(0.6 mmol)的DMAP依次加入上述的混合物中,将所得到的混合物在冰浴下搅拌10 h后,再在室温下搅拌24 h.反应完后,过滤,将滤液旋干后真空干燥,所得到的固体分别用热甲醇和热丙酮洗3−5次后,真空干燥所得到的白色固体即为化合物1,产率为45%.化合物1的表征结果:1H NMR(CDCl3/TMS,300 MHz):δ 7.57(s,2H,benzene),7.45(s,2H,benzene),7.00(s,2H,COONH),5.36(s,2H,alkenyl),5.15(s,2H,CONH),4.46(m,2H,oxycyclohexyl),3.47−3.45(d,4H,NHCH2),3.27−3.26(d,4H,NHCH2),0.68−2.36(m,90H,CH2,cholesteryl protons).元素分析(C72H114N4O6),计算值(%):C,76.18,H,10.05,N,5.08.测定值(%):C,76.07;H,10.17;N,4.74.
2.2.1.3 化合物2的合成
将2.92 g(6 mmol)的化合物a和0.9 mL(6 mmol)三乙胺用150 mL四氢呋喃溶解,将所得混合物在冰浴中搅拌.将0.610 g(3 mmol)间苯二甲酰氯用50 mL四氢呋喃溶解后,在冰浴下慢慢滴加到上述混合物中.滴加完后,将所得混合物在室温下搅拌10 h,将反应混合物过滤,滤液旋干后真空干燥,所得固体分别用热甲醇和热丙酮洗3−5次,真空干燥所得到的白色固体即为化合物2,产率为48%.化合物2的表征结果:1H NMR(CDCl3/TMS,300 MHz):δ 8.28(s,1H,benzene),8.00−7.98(d,2H,benzene),7.50(s,1H,benzene),7.40(s,2H,COONH),5.34(s,4H,alkenyl),5.27(s,2H,CONH),4.47(m,2H,oxycyclohexyl),3.50(s,4H,NHCH2),3.26(s,4H,NHCH2),0.69−2.28(m,90H,CH2,cholesteryl protons).元素分析(C72H114N4O6),计算值(%):C,76.18,H,10.05,N,5.08.测定值(%):C,76.13;H,10.03;N,4.72.
2.2.1.4 化合物3的合成
合成方法同化合物2.化合物3的表征结果:1H NMR(CDCl3/TMS,300 MHz):δ 7.88(d,4H,benzene),7.44(s,2H,COONH),5.37(s,2H,alkenyl),5.14(s,2H,CONH),4.51(m,2H,oxycyclohexyl),3.52(s,4H,NHCH2),3.29(s,4H,NHCH2),0.69−2.28(m,90H,CH2,cholesteryl protons).元素分析(C72H114N4O6),计算值(%):C,76.18,H,10.05,N,5.08.测定值(%):C,76.09;H,10.14;N,4.75.
2.2.2 胶凝实验
将0.025 g胶凝剂与1 mL纯溶剂置于密闭的玻璃样品管中加热至固体完全溶解,室温静置冷却.冷却过程中形成凝胶,记为“G”;冷却过程中部分形成凝胶,记为“PG”;加热过程中胶凝剂基本不溶,记为“I”;胶凝剂在冷却过程中析出沉淀,记为“P”;冷却至室温仍为溶液,记为“S”.
对室温成胶的体系,实验方法为:将0.025 g胶凝剂与1 mL纯溶剂置于密闭的玻璃样品管中,剧烈摇动使胶凝剂充分溶解在溶剂中,室温静置,静置过程中形成凝胶,记为“G(rt)”.
胶凝实验所用有机溶剂均为分析纯,未经纯化直接使用.
2.2.3 结构测定
以扫描电子显微镜(SEM)观察凝胶的形貌,加速电压为15 kV,扫描电流为10 mA;取少量凝胶样品经液氮速冻后冷冻干燥,喷金,进行SEM测定;1H NMR谱的测定以氘代氯仿为溶剂,以TMS为内标;FTIR测定是将固体粉末和干凝胶样品经KBr压片后直接测定,对照样品采用化合物的三氯甲烷稀溶液,将其滴加到KBr片上,干燥后测定;将凝胶样品置于矩形样品池中,冷冻干燥得到干凝胶,进行XRD表征.Cu Kα射线,λ=0.1541 nm,扫描速率为1(°)·min−1.
3 结果与讨论
3.1 化合物1−3的胶凝行为研究
考察了化合物1−3在30种纯溶剂中的胶凝行为,测试浓度为25 mg·mL−1,实验结果列于表1.可以看出,这3种基于胆固醇的A(LS)2型小分子胶凝剂具有不同的胶凝行为.
化合物1能够将30种溶剂中的6种胶凝,其中有3种为芳香烃类溶剂:苯、甲苯和二甲苯.在所测试的11种醇类溶剂中,化合物1能够将己醇和辛醇胶凝,而化合物1在其它醇类溶剂中加热不溶解或形成了沉淀.化合物2能够胶凝5种溶剂,这5种溶剂分别是苯、甲苯、二甲苯、二甲基亚砜(DMSO)和N,N−二甲基甲酰胺(DMF),并且化合物2在室温下就能将DMSO胶凝.
与化合物1和2不同,化合物3的胶凝能力要强的多,它能够将30种被测试溶剂中的24种胶凝,并且包含溶剂种类众多.化合物3不仅能胶凝芳香类溶剂如苯、甲苯和二甲苯等,还能胶凝极性很强的DMSO;不仅能胶凝质子性的醇类溶剂,还能胶凝非质子性的烷烃类溶剂.很明显,具有相似结构的小分子化合物1、2和3表现出了完全不同的胶凝行为.考虑到这3种化合物分子结构的差异,可以认为:胆固醇片断在苯环上相对取代位置的不同对其胶凝行为有极为显著的影响.我们推测,可能是由于化合物3具有完全对称的结构(两个胆固醇片段距离较远),使得其分子结构中的两个胆固醇片段在溶剂中自组装时能够采取更加舒适的方式,从而使其自组装更加容易,因而化合物3的胶凝能力明显优于化合物2和3.

表1 化合物1−3的胶凝行为Table 1 Gelation properties of compounds 1−3
从表1还可以看出,化合物2/DMSO凝胶体系不需要经过加热冷却循环,在室温下就可以直接形成凝胶.同时,化合物3也能在室温下将四氢呋喃、二氯甲烷、四氯化碳和三乙胺这4种有机液体直接胶凝.考虑到室温胶凝的罕见性,以及相关二氯甲烷和四氯化碳与水的互不相溶性,预期依托这些凝胶体系有可能创建新的具有重要应用价值的室温凝胶乳液体系.
与文献12,17上已有的包含大共轭结构的A(LS)2型胆固醇衍生物相比,化合物3能够胶凝的溶剂种类更加广泛,其对芳香类、烷烃类以及醇类溶剂都有一定的胶凝能力,而以苝为A单元的A(LS)2型胆固醇衍生物1a只能将芳香类溶剂如苯、甲苯以及对二甲苯等部分胶凝,但是其对烷烃类和醇类溶剂不具有胶凝能力;12以二苯基丁二烯为A单元的A(LS)2型胆固醇衍生物对醇类溶剂具有较好的胶凝能力,但却不能胶凝烷烃类非极性溶剂.17这表明,利用单个苯环替代大共轭结构作为A单元,显著调节了A(LS)2型胆固醇衍生物的胶凝行为.同时,化合物3与不含A结构的LS2型胆固醇衍生物2b的胶凝行为18相似,这可能是由于单个苯环之间的π−π堆积作用较弱,从而导致含有单个苯环的A(LS)2型化合物与不含苯环的LS2型化合物的胶凝性质相似.
有趣的是,将化合物3在二甲苯中形成的热溶液注入自制的模具中,待体系冷却至室温后,发现形成了化合物3/二甲苯的超分子凝胶薄膜,该薄膜透明且柔韧(如图2所示).在二甲苯存在下,该薄膜可稳定存在.19,20

图2 化合物3/二甲苯凝胶薄膜照片Fig.2 Photographs of the gel film of compound 3/xylene
3.2 凝胶的微观形貌
为了考察胶凝剂分子中苯环上取代基位置的变化对凝胶微观形貌的影响,以SEM考察了不同凝胶体系中胶凝剂分子的聚集结构.图3给出了几个典型凝胶的SEM照片.可以看出,两个胆固醇片段在苯环上取代位置的差异对凝胶的微观形貌有巨大的影响.包含邻位取代结构的化合物1在苯凝胶中形成了互相缠绕的纤维网络结构(图3a),包含间位取代结构的化合物2在苯凝胶中形成的是叶片状结构(图3b),这些“叶片”无序堆积充满凝胶体系,将溶剂固定形成凝胶.而包含对位取代结构的化合物3在苯凝胶中则形成了层状结构(图3c).由此看来,胶凝剂分子结构上的微小差异确实会导致其聚集行为的巨大差异,宏观上表现为胶凝行为、胶凝能力和凝胶性质的差异.
进一步考察图3可以发现,化合物3在苯、甲苯、二甲苯和四氯化碳中形成了完全不同的聚集结构(图3(c−f)).在化合物3/甲苯凝胶中,分子自组装成互相缠绕的纤维网络结构(图3d),而在化合物3/二甲苯凝胶中却形成了孔洞结构(图3e),在化合物3/CCl4中形成的是细纤维堆积成的层状结构(图3f),表明溶剂的性质也显著影响胶凝剂分子的堆积结构.
3.3 红外光谱研究
红外光谱是一种考察氢键是否参与凝胶形成过程的重要手段.因此,本文以化合物1为例,测定了它在不同状态下的FTIR光谱,结果示于图4.图4(a,b)分别对应于化合物1的CHCl3溶液和其苯凝胶的红外光谱.在这里之所以选择用化合物1的CHCl3溶液作为凝胶态的参比是因为CHCl3能很好地溶解化合物1,形成分子水平上的真溶液.考察化合物1/CHCl3溶液的红外光谱可以发现,化合物1的4个典型红外光谱峰为N―H的伸缩振动振动峰,酯基中的C=O伸缩振动峰,酰胺中的C=O伸缩振动峰以及N―H的弯曲振动峰,分别出现在3377、1705、1643和1529 cm−1.然而,在化合物1/苯的干凝胶的红外光谱中看到,这3种典型红外峰的位置分别移动到了3340、1703、1641和1531 cm−1处,这种因氢键的形成和参与导致相应的红外光谱峰发生移动的现象文献19,20已经有过报道.因此,结合胶凝剂分子结构可以推测氢键可能是凝胶形成的重要驱动力之一.

图3 化合物1,2和3在不同溶剂中形成的干凝胶的SEM照片Fig.3 SEM images of the xerogels of compounds 1−3 formed from different solvents
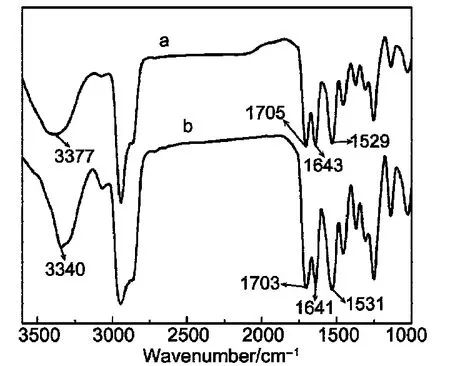
图4 化合物1在CHCl3中(溶液态)(a)及在苯中形成的干凝胶(b)的FTIR光谱Fig.4 FTIR sprectra of compound 1 in CHCl3(solution)(a)and in benzene(xerogel)(b)
3.4 1H NMR研究
为了进一步证实氢键是凝胶形成过程的重要驱动力之一,利用变温和变浓度1H NMR技术研究了化合物1在氘代氯仿中的聚集行为.图5a给出了化合物1在氘代氯仿中的变温1H NMR图谱.很显然,随着体系温度从298 K增加到318 K,两个酰胺质子的化学位移明显向高场移动.在298 K,两个酰胺质子的化学位移分别为7.13和5.27,然而当温度增加到318 K后,这两个酰胺质子的化学位移移动到了7.00和5.15,表明酰胺质子参与了胶凝剂分子氢键的形成.21,22再仔细观察图5a,会发现当温度升高时,除了酰胺质子的化学位移发生了变化外,化合物1苯环质子的化学位移也发生了向低场的微小移动.例如,当温度从298 K增加到318 K,苯环上的质子的化学位移信号分别从7.56和7.46移动到了7.57和7.47.芳香质子的化学位移信号随温度升高而向低场移动的现象可归因于胶凝剂分子中苯环结构发生了π−π堆积.这种胶凝剂分子中芳香基团的π−π堆积作用也可能是推动凝胶形成的重要驱动力.23,24
值得注意的是,温度依赖所发现的氢键作用也可能来自于分子内,如若这样则氢键对凝胶网络形成的作用将会大打折扣.为了确证氢键作用的本源,特别安排了变浓度1H NMR实验,结果示于图5b.可以看出,当化合物1在氘代氯仿中的浓度从10 mg·mL−1增加到25 mg·mL−1时,两个酰胺质子的化学位移信号分别从7.17和5.28移动到了7.20和5.35.同时,苯环上质子的化学位移信号也分别从7.57和7.47移动到了7.54和7.44.两个酰胺质子和苯环上质子的化学位移信号随浓度的变化而发生移动的现象充分说明上述氢键和π−π堆积作用确实源自胶凝剂分子的分子间作用.23,25

图5 化合物1在氘代氯仿中的部分1H NMR图谱Fig.5 Partial1H NMR spectra of compound 1 in CDCl3
3.5 X射线衍射(XRD)分析

图6 化合物1/苯的干凝胶在室温下的XRD图Fig.6 XRD pattern of compound 1/benzene xerogel recorded at room temperature

图7 化合物1在苯中可能的分子堆积模型Fig.7 Probable molecular packing models of compound 1 in benzene
为了揭示胶凝剂分子在凝胶相的分子堆积模式和胶凝机理,26,27利用XRD技术研究了化合物1在苯中形成的干凝胶的堆积结构.为了尽可能保留凝胶中胶凝剂分子的本来聚集结构,用于实验研究的干凝胶样品以快速冷冻和冷冻干燥法制备,所得干凝胶保留了原始新鲜湿凝胶的形貌和体积,外观如同海绵一样,而不是简单固体.XRD测量结果示于图6.从图可以看出,化合物1/苯的XRD曲线包含5个衍射峰,对应的d值分别是4.55、2.56、2.28、1.75和1.28 nm,其对应的比例为,与六方堆积的d值比例)相比较,化合物1/苯干凝胶的衍射峰中除了1/3处的衍射峰未出现外,其余五个峰均符合六方堆积结构.考虑到凝胶的衍射峰本来较弱,可以认为化合1在苯凝胶中形成了六方堆积结构.
根据红外、核磁和XRD研究所得出的胶凝剂分子间的主要相互作用和化合物1/苯凝胶堆积的结构特征,可以尝试提出化合物1在苯凝胶中的可能堆积模型,结果示于图7.分子模拟得到化合物1的分子长度为2.86 nm,六方圆柱体的直径为4.60 nm,这与XRD测定所得到的结果基本一致.
4 结论
合成了3种连接臂包含丙二胺结构的A(LS)2型双胆固醇类小分子胶凝剂并考察了它们在30种常见溶剂中的胶凝行为.实验表明,胆固醇片断在苯环上的取代位置对其胶凝能力影响显著,较之邻位取代的化合物1和间位取代的化合物2,对位取代的化合物3是更有效的胶凝剂.在所发现的胶凝体系中,5种为室温成胶体系.此外,溶剂存在下,化合物3/二甲苯凝胶透明、柔韧,以至于可以形成凝胶薄膜.红外和变温变浓度核磁研究证明除了胆固醇之间的范德华作用外,胶凝剂分子之间的氢键和π−π堆积作用对凝胶的形成也发挥了重要的作用.XRD研究表明化合物1在其苯凝胶中形成六方堆积.
(1)Terech,P.;Weiss,R.G.Chem.Rev.1997,97,313.
(2) Estroff,L.A.;Hamilton,A.D.Chem.Rev.2004,104,1201.doi:10.1021/cr0302049
(3) Sangeetha,N.M.;Maitra,U.Chem.Soc.Rev.2005,34,821.doi:10.1039/b417081b
(4)Ajayaghosh,A.;Praveen,V.K.;Vijayakumar,C.Chem.Soc.Rev.2008,37,109.doi:10.1039/b704456a
(5) George,M.;Weiss,R.G.Accounts Chem.Res.2006,39,489.doi:10.1021/ar0500923
(6)Yang,M.N.;Yan,N.;He,G.;Liu,T.H.;Fang,Y.Acta Phys.-Chim.Sin.2009,25,1040. [杨美妮,晏 妮,何 刚,刘太宏,房 喻.物理化学学报,2009,25,1040.]doi:10.3866/PKU.WHXB20090621
(7)Luo,X.Z.;Wang,Q.;Zhong,J.L.;Pan,H.;Chen,Z.X.Acta Phys.-Chim.Sin.2011,27,1719.[罗序中,王 琼,钟金莲,潘 虹,陈志兴.物理化学学报,2011,27,1719.]doi:10.3866/PKU.WHXB20110720
(8) Gronwald,O.;Snip,E.;Shinkai,S.Curr.Opin.Colloid.In.2002,7,148.doi:10.1016/S1359-0294(02)00016-X
(9) Žinić,M.;Vögtle,F.;Fages,F.Top.Curr.Chem.2005,256,39.doi:10.1007/b105250
(10) Jung,J.H.;Shimizu,T.;Shinkai,S.J.Mater.Chem.2005,15,3979.doi:10.1039/b503441h
(11) Xue,P.;Lu,R.;Li,D.;Jin,M.;Bao,C.;Zhao,C.;Wang,Z.Chem.Mater.2004,16,3702.doi:10.1021/cm034863d
(12) Sugiyasu,K.;Fujita,N.;Shinkai,S.Angew.Chem.Int.Edit.2004,43,1229.
(13) Jung,J.H.;Kobayashi,H.;Masuda,M.;Shimizu,T.;Shinkai,S.J.Am.Chem.Soc.2001,123,8785.doi:10.1021/ja010508h
(14) Xue,M.;Gao,D.;Chen,X.L.;Liu,K.Q.;Fang,Y.J.Colloid Interface Sci.2011,361,556.doi:10.1016/j.jcis.2011.05.074
(15) Xue,M.;Gao,D.;Liu,K.Q.;Peng,J.X.;Fang,Y.Tetrahedron 2009,65,3369.doi:10.1016/j.tet.2009.02.056
(16) Xue,M.;Liu,K.Q.;Peng,J.X.;Zhang,Q.H.;Fang,Y.J.Colloid Interface Sci.2008,327,94.doi:10.1016/j.jcis.2008.08.012
(17) Abraham,S.;Vijayaraghavan,R.K.;Das,S.Langmuir 2009,25,8507.doi:10.1021/la900438c
(18) Peng,J.X.;Liu,K.Q.;Liu,J.;Zhang,Q.H.;Feng,X.L.;Fang,Y.Langmuir 2008,24,2992.doi:10.1021/la703672u
(19) Yoshikawa,I.;Li,J.;Sakata,Y.;Araki,K.Angew.Chem.Int.Edit.2004,43,100.
(20) Liu,J.;He,P.L.;Yan,J.L.;Fang,X.H.;Peng,J.X.;Liu,K.Q.;Fang,Y.Adv.Mater.2008,20,2508.doi:10.1002/adma.v20:13
(21) Shimizu,T.;Masuda,M.J.Am.Chem.Soc.1997,119,2812.doi:10.1021/ja961226y
(22) John,G.;Jung,J.H.;Masuda,M.;Shimizu,T.Langmuir 2004,20,2060.doi:10.1021/la030177h
(23) Wang,C.;Zhang,D.;Zhu,D.Langmuir 2007,23,1478.doi:10.1021/la062621x
(24) Bieser,A.M.;Tiller,J.C.J.Phys.Chem.B 2007,111,13180.doi:10.1021/jp074953w
(25) Jung,J.H.;Shinkai,S.Chem.Eur.J.2002,8,2684.doi:10.1002/1521-3765(20020617)8:12<2684::AID-CHEM2684>3.0.CO;2-Z
(26) Schoonbeek,F.S.;Esch,J.H.;Hulst,R.;Kellogg,R.M.;Fringa,B.L.Chem.Eur.J.2000,6,2633.
(27) Luo,X.Z.;Li,C.;Liang,Y.Q.Chem.Commun.2000,No.2,2091.
