氢原子在Pt及Pt系 双金属催化表面吸附的密度泛函理论研究
2013-09-17高子丰齐随涛伊春海杨伯伦
高子丰 陈 昊 齐随涛 伊春海 杨伯伦
(西安交通大学化工学院化工系,西安710049)
1 引言
氢气作为一种清洁能源的优良载体,具有无污染和高转化效率等诸多优点,因此其制备、分离和储存近年来已成为研究的热点.储氢是氢能大规模利用的关键环节之一.1,2在众多的储氢方法中,有机氢化物储氢具有安全性好,效率高,可以实现大规模、长距离储存与输送等优点,它借助于不饱和芳烃及其对应的有机氢化物之间的可逆加氢和脱氢化学反应实现氢的储存和释放.3−6Pt催化剂被认为是有机氢化物脱氢反应中具有应用前景的催化剂之一,然而由于其昂贵的价格使得工业化成本较高,因此,寻找合适的第二金属掺入Pt既可以降低成本,同时还可能使其具有更好的催化脱氢能力.7−9Nørskov等10认为,催化剂与反应中间体之间应该有适宜的相互作用,不能太强也不能太弱.Chen等11,12在环己烯加氢研究中发现,当催化表面对氢原子的吸附能较高时有利于加氢,而催化表面对氢的吸附能比Pt稍低时,则有利于脱氢.因此,研究氢原子在Pt金属以及Pt系双金属表面的吸附行为,可为脱氢催化剂活性组分的筛选和催化剂结构设计提供一定的理论依据.
随着计算机模拟技术的高速发展,运用密度泛函理论(DFT)计算不同材料的体系能量及电子结构等性质,获取其内在的构效关系,进而实现材料的可控合成已备受关注.目前,研究者们已经对氢吸附在多种过渡金属表面的行为进行了大量探究.Løvvik和Olsen13以及Paul和Sautet14分别计算了氢原子在Pd(111)表面的吸附能,发现氢原子在hcp穴位和桥位的吸附能相近但稳定性明显低于fcc穴位.Kresse和Hafner15研究了氢原子在Ni(100)、(110)和(111)三个表面的吸附情况,结果显示Ni(110)表面能最低.当覆盖度为0.25 ML时,氢原子在这三种表面最稳定吸附位均是fcc穴位.黄永丽和刘志平16研究了氢和硫原子在金属Pd、Au、Cu表面的吸附,结果表明氢原子在此三种金属(111)表面的最稳定吸附位均为fcc穴位,氢原子在Pd表面吸附最稳定,Cu次之,Au最差.Watson等17研究了氢原子在Ni、Pd、Pt金属(111)表面的吸附情况,发现Ni和Pd上氢原子的穴位吸附比顶位吸附更为稳定,而Pt上氢原子在不同吸附位的吸附能相近,使其表面扩散较为容易.此外,Lima等18采用实验和DFT结合的方法,研究了Pt(111)面掺杂3d过渡金属构成的双金属表面催化活性与氢原子吸附行为的内在联系,验证了利用DFT进行催化材料设计的可靠性.
本文基于密度泛函理论,探讨了Pt(100)、(110)、(111)三种不同表面的表面能、层间弛豫量以及氢原子在不同Pt表面上的吸附能和相关键长变化的情况,考察了掺杂第二金属后的M-Pt(111)双金属(M=Al,Fe,Co,Ni,Cu,Pd)表面的氢原子吸附行为及吸附前后双金属表面的层间弛豫情况,最后通过对氢原子在双金属表面吸附前后的局域态密度以及双金属表面偏离费米能级程度综合分析,并与氢原子吸附强弱进行关联以预测不同双金属表面的催化脱氢活性.
2 理论模型和计算方法
密度泛函计算均采用了维也纳从头计算模拟程序包(简称VASP).电子交换相关部分在广义梯度近似(GGA)下用标准PBE(Perdew-Burke-Ernzerhof)交换关联泛函进行描述.计算过程采用p(2×2)的结构模型,在z方向周期性排列的相邻表面层之间留有厚度为1 nm的真空区域以避免表面间的相互干扰.鉴于金属Pt的高指数台阶面大多是由(100)、(110)、(111)三种低指数表面构成,19研究中选取金属Pt(100)、(110)、(111)表面进行弛豫计算.M-Pt(111)双金属表面结构模型均基于Pt(111)表面,将第一层Pt原子用相应的第二金属取代.H原子在Pt及M-Pt(111)双金属表面的模拟计算均选择覆盖度为0.25 ML.
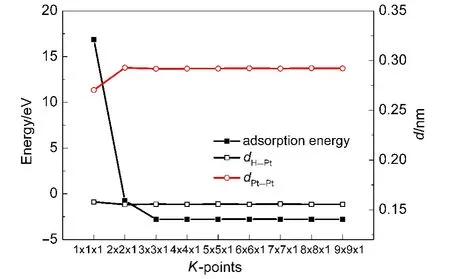
图1 不同K点密度下的氢吸附能及H―Pt、Pt―Pt键长Fig.1 Hydrogen adsorption energies and the bond lengths of H―Pt,Pt―Pt under the condition of different K-point densities
研究中采用的四层(或六层)结构模型,在计算金属表面的弛豫和H原子吸附时,只允许上两层(或上三层)Pt原子在平衡位置附近弛豫,下两层(或下三层)原子均固定不动.原子结构优化中的总能收敛性判据设定为10−5eV,平面波截断能取为400 eV.选取H原子在Pt(111)的顶位吸附对二维布里渊区(Brillouin zone)K点网格密度进行收敛性验证.以Γ点为中心,采用Monkhorst-Pack方案自动产生不可约K点作自恰计算,分别从1×1×1顺次增加到9×9×1,验证结果如图1所示.当K点密度取3×3×1时,吸附能偏差即可控制在0.005 eV以内,计算所得H―Pt键长以及层间Pt―Pt键长偏差很小,在该条件下优化得到Pt的晶格常数为0.394 nm,与实验值0.392 nm较为吻合,20表明上述设置条件足以保证计算结果的准确性.
3 计算结果与讨论
3.1 Pt表面结构模型优化
研究中分别选取Pt(100)、(110)、(111)三种表面的四层和六层结构模型以考察不同表面及不同层数的结构弛豫情况,探讨能准确模拟表面性质的模型层数.金属表面能(Eσ)计算遵循公式(1).

其中,Eslab是优化后衬底的总能量;Ebulk是单个原子的能量;N是超晶胞内的原子总数;A是表面截面积.表1和表2列出了计算得到的表面能和表面弛豫前后层间Pt―Pt键长变化结果.弛豫后不同Pt表面能的大小顺序为Eσ(111)<Eσ(100)<Eσ(110),Pt(111)的表面能最低.相比于四层模型,六层模型的表面能高大约0.1−0.2 J·m−2.将上述结果与文献21,22的计算结果比较,发现结果整体变化趋势相同,证实了上述计算的可靠性.对比三个不同Pt表面弛豫层间的Pt―Pt键长发现,四层模型中Pt(100)和Pt(110)面相对于原胞均有不同程度的内缩,且最外层Pt内缩量最大,分别为1.42%和3.05%,但Pt(111)面的弛豫量较小,仅为0.25%.六层模型中Pt(100)和Pt(110)的弛豫层向内收缩程度增大,而Pt(111)的第一、二层,第二、三层和第三、四层的层间Pt―Pt键长相差不大,最外层的弛豫量只有0.04%,同原胞的Pt―Pt键长非常接近,表明Pt(111)表面有很好的稳定性.采用六层和四层模型计算所得的Pt(111)表面上H原子的顶位吸附能分别为−2.74和−2.78 eV.就两种不同层数模型结果比较而言,二者在表面能、层间Pt―Pt键长和H原子的吸附能结果相差很小,表明四层模型已足够用来模拟金属的表面性质,因此后续计算均采用四层模型.

表1 不同层数Pt表面的表面能(Eσ)Table 1 Surface energies(Eσ)of Pt surfaces with different layers
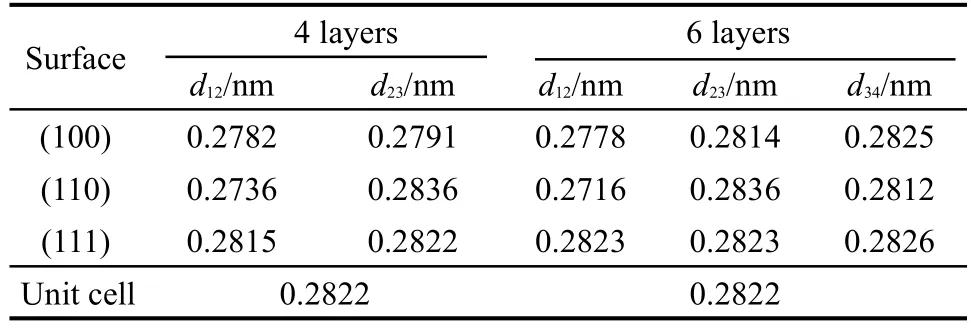
表2 不同层数Pt表面弛豫后的Pt―Pt键长Table 2 Bond lengths of Pt―Pt after relaxation with different layers
3.2 Pt三种表面及M-Pt(111)双金属表面上H原子的吸附
图2是Pt(100)、(110)、(111)表面上H原子吸附位置示意图.H原子在Pt(100)表面有顶位(T)、桥位(B)、穴位(H)三种吸附位置,在Pt(110)表面和Pt(111)表面均有四种吸附位置,分别为顶位(T)、长桥位(BL)、短桥位(BS)、穴位(H)和顶位(T)、桥位(B)、hcp穴位、fcc穴位.

图2 Pt(100)、(110)、(111)表面上H原子的吸附位置Fig.2 Adsorption sites of H atoms on Pt(100),(110),(111)surfaces
H原子在金属表面的吸附能(EA)按照公式(2)进行计算,其中,EH+slab是金属衬底吸附H原子后总能量;EH是H原子的能量.

不同Pt表面上H原子的吸附模型优化后的计算结果列于表3.在Pt(100)上,H原子最易为桥位吸附,其次为顶位和穴位吸附.在Pt(110)上,H原子最易为短桥位吸附,随后是顶位及长桥位吸附,穴位吸附最难.相比而言,Pt(111)上H原子在四个不同位置的吸附能差别很小且均很低,表明该表面上H原子从一处移动到另一处所需能量较小,有利于表面上的扩散.其中,H原子最易为fcc穴位吸附,其次是hcp穴位和桥位、顶位吸附,此结果与马淳安等23的结论一致.
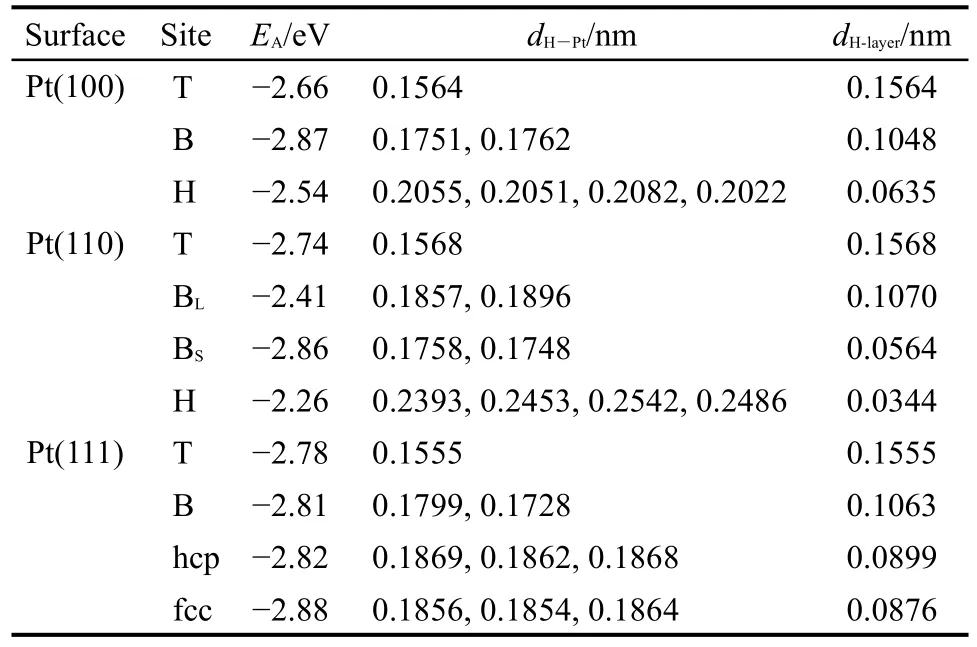
表3 H在Pt表面不同位置吸附的计算结果Table 3 Calculation results of H adsorption at different sites of Pt surfaces
为了考察第二金属掺杂后对H原子吸附行为的影响,研究中选取Al、Fe、Co、Ni、Cu、Pd等作为第二金属.图3和表4分别为M-Pt双金属(111)表面上H原子的吸附模型示意图和计算结果.所有双金属催化剂表面上,H原子最可能吸附位均为fcc穴位.其中,H原子在Ni-Pt和Co-Pt双金属表面上不同位置的吸附能均很接近,在Ni-Pt双金属表面的fcc穴位吸附能最低,为−3.13 eV,随后为桥位和hcp穴位吸附,在顶位的吸附能较高.在Fe-Pt双金属表面,H原子在fcc穴位和hcp穴位的吸附能分别为−3.03和−2.93 eV,但在顶位和桥位吸附能却为−1.71和−0.87 eV,均高于H原子的平均结合能−2.27 eV,表明H原子优先吸附在两个穴位上,而不会以原子形式吸附在该双金属的顶位和桥位.在Pd-Pt双金属上,H原子以fcc穴位的吸附能为最低,但却高于纯Pt上fcc穴位的H原子吸附能.当采用主族元素Al作为掺杂金属时,H原子在Al-Pt表面上的最佳吸附位亦为fcc穴位,对应的吸附能为−2.42 eV,其他三个位置的吸附能均高于H原子的平均结合能,表明H原子在Al-Pt双金属催化剂表面吸附能力较差,更易从该催化剂表面脱附而结合形成H2分子.H原子在Cu-Pt双金属表面的各个吸附位的吸附行为与Al-Pt类似.
图3 M-Pt双金属(111)表面上H原子的吸附模型Fig.3 Adsorption model of H atom on M-Pt(111)bimetallic surfaces

表4 H在M-Pt(111)双金属表面不同位置吸附的计算结果Table 4 Calculation results of H adsorption at different sites of M-Pt(111)bimetallic surfaces
3.3 Pt(111)及M-Pt(111)双金属表面上吸附H原子后的表面弛豫分析
金属表面的弛豫现象可反映出吸附物种与金属表面的相互作用情况,表5为Pt及Pt系双金属(111)表面H原子在fcc穴位吸附后的金属表面弛豫量计算结果.正弛豫量表示吸附H原子后金属原子层间距变大,弛豫层向外膨胀,表明该双金属表面与H原子作用较强,反之亦然.吸附H原子后的Pt(111)面,其第一、二层原子间距变大,第二、三层间距稍有变小,而Ni、Co、Fe掺杂的双金属表面的弛豫量很接近,第一、二层和第二、三层间距均有不同程度变大,表明此三种金属在吸附过程中外层的第二金属原子向外膨胀,与H原子有较强的吸附作用.Pd-Pt双金属表面虽有较大的弛豫量,但相对于前三种双金属,其第一、二层原子间距变化幅度较小,而第二、三层变化幅度较大,表明该表面在吸附H原子时,第二层Pt原子向外膨胀,对H原子吸附有显著贡献.Al-Pt、Cu-Pt这两种双金属表面,第一、二层原子的间距在吸附H原子后减小,而第二、三层Pt原子间距则变大,说明表层第二金属Al和Cu本身对于H原子吸附较弱,而更倾向于同第二层的Pt相互作用,较好解释了Pt掺杂该两种金属后对于H原子吸附作用并未得到改善的原因.
3.4 电子态密度分析
电子态密度可反映原子轨道上的电子能量变化,进而揭示出吸附过程中的成键状态.为了验证H原子在Pt(111)及M-Pt(111)双金属表面吸附能的计算结果,对H原子在不同金属表面吸附前后的电子状态进行了自洽计算并得到相应的局域态密度,计算过程中费米能级被定为能量零点.
图4为H原子在不同双金属表面吸附前后的局域态密度图.其中图4(a)是未吸附在金属表面的H原子态密度,可以看出H原子在费米能级处有很强的峰.图4(e)表明当吸附在Pt表面时,H原子的离域性变强,证实了H原子与Pt原子形成了化学键.将图4(b,c,d)与图4(e)进行对比发现,在Ni-Pt、Co-Pt、Fe-Pt表面吸附着的H原子离域性更强,所形成的化学键更加稳固.然而,由图4(f,g,h)与图4(e)的对比可知在Pd-Pt、Cu-Pt、Al-Pt表面吸附着的H原子离域性相对较差,H原子与金属原子所形成的化学键较弱.通过态密度图得出来的结论与之前H原子吸附能的计算结果吻合.
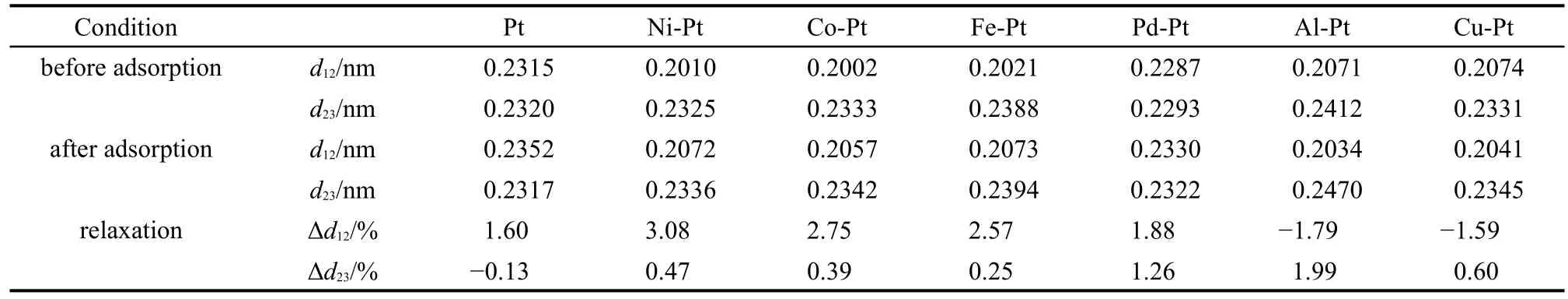
表5 Pt及Pt系双金属(111)表面上fcc穴位吸附H原子后的表面弛豫量(Δd)Table 5 Amount of surface relaxation(Δd)after hydrogen adsorbed at fcc hollow site on Pt and M-Pt(111)bimetallic surfaces

图4 不同M-Pt(111)双金属表面上H原子的局域态密度Fig.4 Local density of states of H atom on different M-Pt(111)bimetallic surfaces
3.5 Pt(111)及M-Pt(111)双金属表面d带中心与氢原子吸附强弱的关联
对于Pt及Pt系双金属表面,d带电子对总态密度的贡献最大.d带中心可较好地反映出过渡金属表面的电子特性,并且与小分子的吸附能存在一定关联,据此可对不同金属表面吸附H原子能力进行有效验证和预测.24,25第二金属的掺杂使得双金属表面的电荷密度会有不同程度的变化,进而使得d带变宽或变窄,相应地,d带中心就会远离或靠近费米能级.不同M-Pt(111)双金属表面上的H原子吸附能大小与其d带中心的关联如图5所示.Ni、Co、Fe等3d金属的掺杂,使得3d-Pt(111)双金属表面的d带中心发生迁移,较纯Pt更靠近费米能级.这是由于此三种过渡金属的d带均为不饱和状态,从而与H原子s带电子存在较强的相互作用,促使相应的双金属表面对H原子的吸附能力加强.Pd-Pt和Cu-Pt的d带中心较Pt远离费米能级,其表面H原子吸附能也相对较高,是因为Pd和Cu的d带均为满电子状态,因此呈现出一种排斥H原子s带电子的趋势,对H原子的吸附性较弱.Al-Pt双金属表面的d带中心较Pt偏移很小,对H的吸附效果相对较差.综上分析发现,第二金属的掺杂通过其d带电子作用使MPt(111)双金属表面d带中心产生偏移,其偏离费米能级的程度与H原子在金属表面的吸附能大小存在一定的关联,即金属表面d带中心越靠近费米能级,则其吸附H原子的能力越强,可能具有更好的催化脱氢活性.
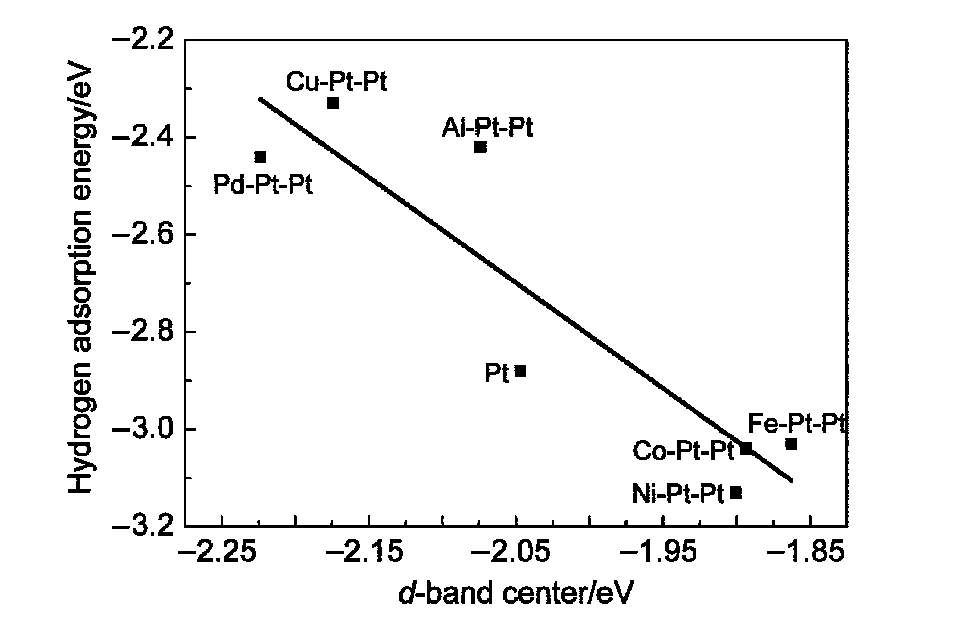
图5 H原子吸附能与M-Pt(111)双金属表面d带中心的关联Fig.5 Correlation between hydrogen adsorption energies and d-band centers of M-Pt(111)bimetallic surfaces
4 结论
采用密度泛函理论,探讨了覆盖度为0.25 ML时H原子在Pt三种表面不同位置的吸附作用,分析了第二金属掺杂的M-Pt(111)双金属对H原子的吸附情况、吸附后的金属层间弛豫变化,并进一步将金属表面d带中心与对氢原子的吸附强弱进行了关联.所得结论如下:
(1)Pt三种表面中以Pt(111)面最稳定,H原子在Pt(111)表面各个位置的吸附能差别很小,且均为稳定吸附.
(2)H原子在第二金属掺杂的M-Pt(111)双金属表面最可能吸附位均为fcc穴位.其在Ni-Pt双金属表面的吸附能最低为−3.13 eV,Co-Pt和Fe-Pt次之,分别为−3.04 eV和−3.03 eV.H原子吸附前后局域态密度的变化能定性地反映其吸附能的相对强弱.
(3)3d-Pt(111)双金属表面在吸附H原子后,第一层金属原子向外扩张明显,第二层金属原子也有不同程度的外扩倾向;Pd-Pt双金属弛豫层外扩量也较大,但其第一层原子外扩的幅度比前三种双金属要小;Al-Pt和Cu-Pt双金属表面的第一、二层原子间距在吸附H原子后减小,而第二、三层Pt原子间距变大.H原子的吸附能大小与不同M-Pt(111)双金属表面的d带中心位置间存在一定关联,d带中心越靠近费米能级,H原子的吸附能力越强.
(1) Murray,L.J.;Dincă,M.;Long,J.R.Chem.Soc.Rev.2009,38(5),1294.doi:10.1039/b802256a
(2) Shukla,A.A.;Gosavi,P.V.;Pande,J.V.;Kumar,V.P.;Chary,K.V.;Biniwale,R.B.Int.J.Hydrog.Energy 2010,35(9),4020.doi:10.1016/j.ijhydene.2010.02.014
(3) Bhasin,M.M.;McCain,J.H.;Vora,B.V.;Imai,T.;Pujado,P.R.Appl.Catal.A 2001,221(1),397.
(4) Qi,S.T.;Yu,W.T.;William,W.L.;Yang,B.L.;Chen,J.G.Chin.J.Catal.2010,31(8),955.[齐随涛,俞伟婷,William W.Lonergan,杨伯伦,陈经广.催化学报,2010,31(8),955.]doi:10.1016/S1872-2067(09)60092-9
(5) Iwasa,N.;Takezawa,N.Top.Catal.2003,22(3−4),215.
(6) Zhu,G.L.;Yang,B.L.Prog.Chem.2009,21(12),2760.[朱刚利,杨伯伦.化学进展,2009,21(12),2760.]
(7) Desai,S.K.;Neurock,M.;Kourtakis,K.J.Phys.Chem.B 2002,106(10),2559.doi:10.1021/jp0132984
(8) Flick,D.W.;Huff,M.C.Appl.Catal.A 1999,187(1),13.doi:10.1016/S0926-860X(99)00179-9
(9) Qi,S.T.;Huang,J.;Chen,H.;Gao,Z.F.;Yi,C.H.;Yang,B.L.Acta Chim.Sin.2012,70(24),2467. [齐随涛,黄 俊,陈 昊,高子丰,伊春海,杨伯伦.化学学报,2012,70(24),2467.]doi:10.6023/A12080603
(10) Nørskov,J.K.;Rossmeisl,J.;Logadottir,A.;Lindqvist,L.;Kitchin,J.R.;Bligaard,T.;Jonsson,H.J.Phys.Chem.B 2004,108(46),17886.doi:10.1021/jp047349j
(11) Chen,J.G.;Menning,C.A.;Zellner,M.B.Surf.Sci.Rep.2008,63(5),201.doi:10.1016/j.surfrep.2008.02.001
(12) Chen,J.G.;Qi,S.T.;Humbert,M.P.;Menning,C.A.;Zhu,Y.X.Acta Phys.-Chim.Sin.2010,26(4),869.[陈经广,齐随涛,Humbert,M.P.,Menning,C.A.,朱月香.物理化学学报,2010,26(4),869.]doi:10.3866/PKU.WHXB20100441
(13) Løvvik,O.M.;Olsen,R.A.Phys.Rev.B 1998,58(16),10890.doi:10.1103/PhysRevB.58.10890
(14) Paul,J.F.;Sautet,P.Phys.Rev.B 1996,53(12),8015.doi:10.1103/PhysRevB.53.8015
(15) Kresse,G.;Hafner,J.Surf.Sci.2000,459(3),287.doi:10.1016/S0039-6028(00)00457-X
(16) Huang,Y.L.;Liu,Z.P.Acta Phys.-Chim.Sin.2008,24(9),1662.[黄永丽,刘志平.物理化学学报,2008,24(9),1662.]doi:10.3866/PKU.WHXB20080923
(17) Watson,G.W.;Wells,R.P.;Willock,D.J.;Hutchings,G.J.J.Phys.Chem.B 2001,105(21),4889.doi:10.1021/jp002864c
(18) Lima,F.H.;Zhang,J.;Shao,M.H.;Sasaki,K.;Vukmirovic,M.B.;Ticianelli,E.A.;Adzic,R.R.J.Phys.Chem.C 2007,111(1),404.doi:10.1021/jp065181r
(19) Ren,Y.P.;Lu,Y.X.;Zi,Q.Acta Phys.-Chim.Sin.2007,23(11),1728.[任云鹏,鲁玉祥,姿 琦.物理化学学报,2007,23(11),1728.]doi:10.3866/PKU.WHXB20071114
(20) Lynch,M.;Hu,P.Surf.Sci.2000,458(1),1.
(21) Zhang,J.M.;Ma,F.;Xu,K.W.Appl.Surf.Sci.2004,229(1),34.
(22) Vitos,L.;Ruban,A.;Skriver,H.L.;Kollar,J.Surf.Sci.1998,411(1),186.
(23) Ma,C.A.;Liu,T.;Chen,L.T.Acta Phys.-Chim.Sin.2010,26(1),155.[马淳安,刘 婷,陈丽涛.物理化学学报,2010,26(1),155.]doi:10.3866/PKU.WHXB20091224
(24) Vegge,T.;Hedegaard-Jensen,L.S.;Bonde,J.;Munter,T.R.;Nϕrskov,J.K.J.Alloy.Compd.2005,386(1),1.
(25) Bligaard,T.;Nørskov,J.K.Electrochim.Acta 2007,52(18),5512.doi:10.1016/j.electacta.2007.02.041
