活性炭基软包装超级电容器用有机电解液
2013-09-21孙现众张大成马衍伟
黄 博 孙现众 张 熊 张大成 马衍伟
(中国科学院电工研究所应用超导重点实验室,北京100190)
1 引言
超级电容器(supercapacitors或ultracapacitors)也叫电化学电容器,1−4因其具有适中的能量密度和功率密度、较长的循环寿命、绿色环保等优点,在新能源发电、电动汽车、信息技术、航空航天和国防等领域都有着广阔的应用前景.5−10超级电容器电极材料主要分为碳材料、金属氧化物、导电聚合物等.目前,在很多文献中,超级电容器专指以碳材料为电极材料的电化学双电层电容器,本文亦沿用这一用法.超级电容器靠活性物质与电解液的界面存储电荷,11没有法拉第反应发生,12其存储的能量E=1/2·CU2.由此可知,电容器的能量密度E与比电容C和工作电压U的平方均成正比.4因此可以从提高电极材料比电容和工作电压两方面入手来提高超级电容器的比能量.13超级电容器的比电容往往由电极材料和电解液共同决定,14而电压窗口主要由电解液决定.因此,电解液对超级电容器的比能量有着决定性的作用.
目前所使用的超级电容器电解液有水系电解液、有机电解液和离子液体.水系电解液多为离子导电性较好的硫酸、氢氧化钾等溶液,比电容高但电压窗口窄(只有1 V左右),比能量较低,因而其产业化受到制约.有机电解液电压窗口较宽(可达2−4 V),比能量较高,是当今的研究热点.离子液体主要由二(三氟甲基磺酰)亚胺(TFSI−)、BF4−和 PF6−等阴离子与咪唑类、吡咯类及短链脂肪季胺盐类等有机阳离子构成,7它具有不易挥发、热稳定性好及电化学窗口宽等特点,15−17但是离子液体也存在诸多缺点,如明显较低的比电容、较高的粘度和内阻.17,18
目前商品化双电层电容器常用的电解液为1 mol·L−1四乙基四氟硼酸铵的乙腈溶液(Et4NBF4/AN)和1 mol·L−1四乙基四氟硼酸铵的丙烯碳酸酯溶液(Et4NBF4/PC).乙腈为非质子极性溶剂,粘度低但易挥发.而丙烯碳酸酯是一种强极性溶剂,熔点低,沸点高但粘度偏高.因此,采用乙腈和丙烯碳酸酯混合溶剂发挥各自的特点,将有助于提高电解液的稳定性和电压窗口.19−21
本文采用活性炭作为电极极化活性物质,组装成软包装超级电容器,首次研究了其在1 mol·L−1三乙基甲基四氟硼酸铵(MeEt3NBF4)的AN和PC混合溶剂(质量比为1:1)中的电化学性能.MeEt3NBF4的溶解度和电化学分解电压比Et4NBF4高,此外还具有阴阳离子半径大小相当等特点.我们分别以(1 mol·L−1MeEt3NBF4/(AN+PC)、1 mol·L−1Et4NBF4/AN和1 mol·L−1Et4NBF4/PC)为电解液制作活性炭基超级电容器,并在3 V电压下对它们的循环伏安、电化学阻抗谱、恒流充放电、漏电流、自放电、循环寿命和库仑效率等电化学性能进行了系统比较.
2 实验
2.1 实验材料
本文使用的新型有机电解液由北京化学试剂研究所提供,电导率为40 mS·cm−1,溶剂为乙腈和丙烯碳酸酯按1:1的质量比混合而成(电池级),电解质为MeEt3NBF4,记为ME;1 mol·L−1Et4NBF4/AN溶液记为 AN(溶剂纯度99.9986),1 mol·L−1Et4NBF4/PC溶液记为PC(溶剂纯度99.9974),电导率分别为55.21和13.35 mS·cm−1,以上两种电解液均来自于深圳新宙邦公司.使用导电炭黑(super C45,SC)作导电剂.粘接剂是羧甲基纤维素钠(CMC)和丁苯橡胶(SBR)按1:2质量比混合而成,使用刻蚀铝箔(JCC,20 μm)作为集流体以减少铝箔与活性物质之间的接触电阻.22
2.2 超级电容器制备方法
电极片的制备方法如下,按质量比m(AC):m(SC):m(CMC):m(SBR)=80:10:2:4的比例备料,将CMC和SBR溶于去离子水中,磁力搅拌1 h得到均匀的CMC和SBR混合溶液备用.将活性炭、导电炭黑球磨30 min混匀,加入已制备好的CMC和SBR混合溶液后再球磨2 h混匀.将混匀的浆料用自动涂布机均匀地涂在铝箔上,涂布厚度为100 μm.然后,转移至干燥箱中,在空气气氛下,80°C干燥12 h.
超级电容器制备方法为:将极片冲成3.5 cm×4 cm的矩形.再用压片机以相同的压力将极片压实,用胶带将极片和隔膜按正极-隔膜-负极的顺序裹好.用超声点焊机在极耳处焊接上引出电极,置于铝塑包内,然后100°C真空干燥24 h.快速转移至氩气手套箱(水分含量和氧气含量均小于1.0×10−7)中,分别注入三种不同的电解液,移出手套箱,真空封口.
2.3 电化学测试
在上海辰华公司CHI660c电化学工作站上进行循环伏安(CV)测试.使用Autolab对其进行电化学阻抗谱(EIS)测试,交流信号振幅为10 mV,频率范围为0.01 Hz−100 kHz.充放电实验在美国Arbin公司MSTAT4电化学测试系统上完成.漏电流测试方法为先以1 A·g−1(质量按正负极极化活性物质AC总质量计算,下同)的电流密度,恒流充至额定电压,再以额定电压恒压充电2 h后,测得的电流作为漏电流,漏电流测试同样在Arbin电化学测试系统上完成.循环性能测试在武汉Land电化学测试系统上进行,充放电电流密度为1A·g−1.
特制三电极体系是在两电极体系的基础上,在工作电极和对电极的外侧各添加一片新的AC电极,然后将两片新的AC电极通过同一引出电极引出作为参比电极,AC参比电极用隔膜与工作电极和对电极隔开.
超级电容器的电极材料比电容Cs(单位:F·g−1)、内阻R(单位:Ω)和库仑效率η可以利用恒流充放电实验,按以下公式求得.23

其中:I为恒流充放电电流(单位:A),M为正负极极化活性物质(AC)总质量(单位:g),Δt为放电时间(单位:s),ΔU为充放电电流换向时的电压降(单位:V),U∆为电压窗口上限与ΔU之差(单位:V),tdis为放电时间(单位:s),tch为充电时间(单位:s).
3 结果和讨论
3.1 循环伏安
循环伏安曲线可用来判断超级电容器的内阻特性,表征超级电容器的容量特性和确定电解液的电压窗口.对于理想的电容行为,CV曲线呈现出对称的矩形,由于超级电容器的内阻,CV曲线的矩形形状将发生扭曲(矩形的两竖线变成弯曲的斜线),内阻越大,扭曲越严重.由于矩形的面积与充电容量和放电容量之和成正比,所以,矩形面积越大说明超级电容器的容量越高.同时当CV曲线电流出现较快增长时,表明超级电容器内电解液开始分解.
图1(a)是ME电解液在不同电压窗口下的CV曲线,扫描速率是20 mV·s−1,纵坐标为充放电电流(单位:A).从图中可以看出,在2.5−3.1 V的截止电压下,电流无明显增加,同时CV曲线的扭曲程度未明显加强,说明超级电容器的内阻无明显增大并且ME电解液在3.1 V下未发生明显分解.在3.3和3.5 V下,电流有明显提高,同时扭曲程度明显加强,说明电解液分解程度更加严重,分解产物使超级电容器的内阻开始增大.
图1(b)是三种超级电容器在3 V下的CV曲线,扫描速率是20 mV·s−1,纵坐标为电流密度(充放电电流与极化活性物质总量之比,单位:A·g−1).从图中可以看出,AN和ME电解液的CV曲线大致重合,这说明AN和ME电解液有非常相似的电容特性.同时,AN和ME电解液较PC的CV曲线扭曲程度更小,这说明AN和ME电解液的内阻更低.由于PC相对严重的扭曲,其CV曲线的面积要小于AN和ME电解液,可以得知PC下的比电容将不及AN和ME下的比电容,并且AN的CV曲线面积比ME电解液的CV曲线面积稍大,可知,AN的比电容较ME电解液稍大.
图1(c)是ME电解液在不同扫描速率(20、50、100、200 mV·s−1)下的CV曲线,电压窗口为0−3 V.可见,ME电解液在200 mV·s−1的扫描速率下依然能够保持较好的矩形形状,可知,在这一电解液下有较好的倍率特性,这归功于这种电解液有较高的电导率(40 mS·cm−1).

图1 超级电容器的循环伏安(CV)曲线Fig.1 Cyclic voltammetry(CV)curves of supercapacitors
3.2 电化学阻抗谱
电化学阻抗谱(EIS)的测量方法是,通过施加某一频率ω的正弦电压 ∆E(ω)=∆Emaxejωt, 得到同频率ω下的正弦电流 ∆I(ω)=∆Imaxejωt-φ,输出信号 ∆E(ω)与相应输入信号∆I(ω)的比值就是在该频率ω下的交流阻抗:其中,″分别是Z(ω)的实部和虚部.
使用不同电解液的超级电容器的电化学阻抗谱如图2(a)所示,振幅均为10 mV,频率为0.01 Hz−100 kHz.由图可见,三种超级电容器的EIS形状相似,主要由高频区的半圆弧、中频区的45°斜线和低频区的竖线三部分组成.高频区的半圆弧与电荷转移阻抗和极化阻抗有关.24,25中频区的45°斜线主要由多孔活性炭电极的分布电容和分布电阻引起,低频区的竖线由超级电容器的双电层电容引起,竖线越接近竖直,说明电容的电容性越好.曲线与实轴的交点被称为溶液电阻RE,其主要是电解液的内阻,另外还包括了活性炭电极的内阻、活性炭电极与集流体间的接触电阻.半圆弧所引起的电阻增加部分被称为电荷转移电阻Rct.电荷转移电阻与溶液电阻一起构成等效串联电阻(ESR).活性炭多孔结构里的电解液电阻被称为等效分布电阻(EDR),26低频区竖线的延长线与实轴的交点被称为超级电容器的总内阻(Rtotal),Rtotal=ESR+EDR.
由图2可知,三种电容的总内阻Rtotal按PC、ME、AN的顺序依次减小,同时溶液电阻RE和EDR也按PC、ME、AN的顺序依次减小,这与三种电解液的电导率大小是相吻合的,ME电解液电导率居中,符合商用电解液电导率要求.PC电解液有较低的电导率原因在于其较高的粘度.27,28同时,在低频区,三条斜线相互平行,说明三种电容的电容特性接近.
图2(b)是ME电解液在不同截止电压下充放电后的EIS,测试方法为:将超级电容器依次在递增的截止电压下充放电,每一截止电压下充放电5周后,测试超级电容器的EIS.由图可见,随着截止电压的提高,超级电容器的电荷转移电阻Rct不断增大,这主要是由电解液在较高截止电压下的分解产物所引起.同时电荷转移电阻Rct的增长速度也在增大,这说明超级电容器的电解液分解加快.例如,截止电压从2.5 V提高到2.6 V时的Rct增量要明显小于电压从3 V提高到3.1 V的Rct增量.

图2 超级电容器的电化学阻抗谱(EIS)和电荷转移电阻(Rct)Fig.2 Electrochemical impedance spectroscopy(EIS)and charge transfer resistance(Rct)of supercapacitors
为了定量分析Rct增量变化情况,用Zview软件求出图2(b)中Rct,图2(c)是超级电容器在不同截止电压下充放电后的电荷转移电阻Rct大小.由图可知,电荷转移电阻Rct随截止电压的增大而不断增大,同时曲线的斜率随截止电压的增大而不断增大,说明随着截止电压的提高,电解液的分解程度越来越严重,尤其是在3.3和3.5 V下,电荷转移电阻Rct增大十分明显,这与ME电解液在不同截止电压下的EIS和CV曲线取得很好的一致性.
3.3 恒流充放电和比电容
图3(a)为ME电解液在不同电流密度(相对于正负极AC总质量)下的恒流充放电曲线,电流密度分别为0.2、0.5、1、2、5、10 A·g−1,电压窗口为0−3 V.根据公式(1)-(3)可计算出三种超级电容器在不同电流密度下的比电容Cs、内阻R.经计算,等效内阻R随电流密度的变化不大,但不同电容的内阻不同,ME电解液为0.204 Ω,AN为0.176 Ω,PC为0.301 Ω,这表明ME电解液电导率居中,这与三者的EIS和电导率是一致的.
图3(b)是3 V下三种超级电容器的比电容Cs随电流密度的变化关系,由图可知,在200 mA·g−1的电流密度下,三种超级电容器ME、AN和PC的比电容分别为144.70、145.92 和142.03 F·g−1,AN的比电容最高,PC的比电容最低,这主要是因为AN的分子较PC分子小,所以AN的溶剂化离子比PC的溶剂化离子更小,更易深入活性炭多孔电极的微孔.在电流密度低于2 A·g−1时,三种超级电容器的比电容均快速下降.随着电流密度的继续增大,ME和AN的下降趋势十分相似,下降程度要明显低于PC.
图3(c)是三种电解液下三电极体系超级电容器在3V下的充放电曲线,充放电电流密度为0.2A·g−1.可见,放电时间AN>ME>PC,即比电容AN>ME>PC,ME的比电容居中,AN的比电容比PC大,还可以看出,三种超级电容器的正极的电位窗口比负极的电位窗口窄,由此可知,三种电解液下,正极的比电容比负极的比电容大,经计算ME、AN、PC三种超级电容器的正负极比电容之比分别为:1.109、1.088、1.149,ME电解液有适中的正负极比电容比.
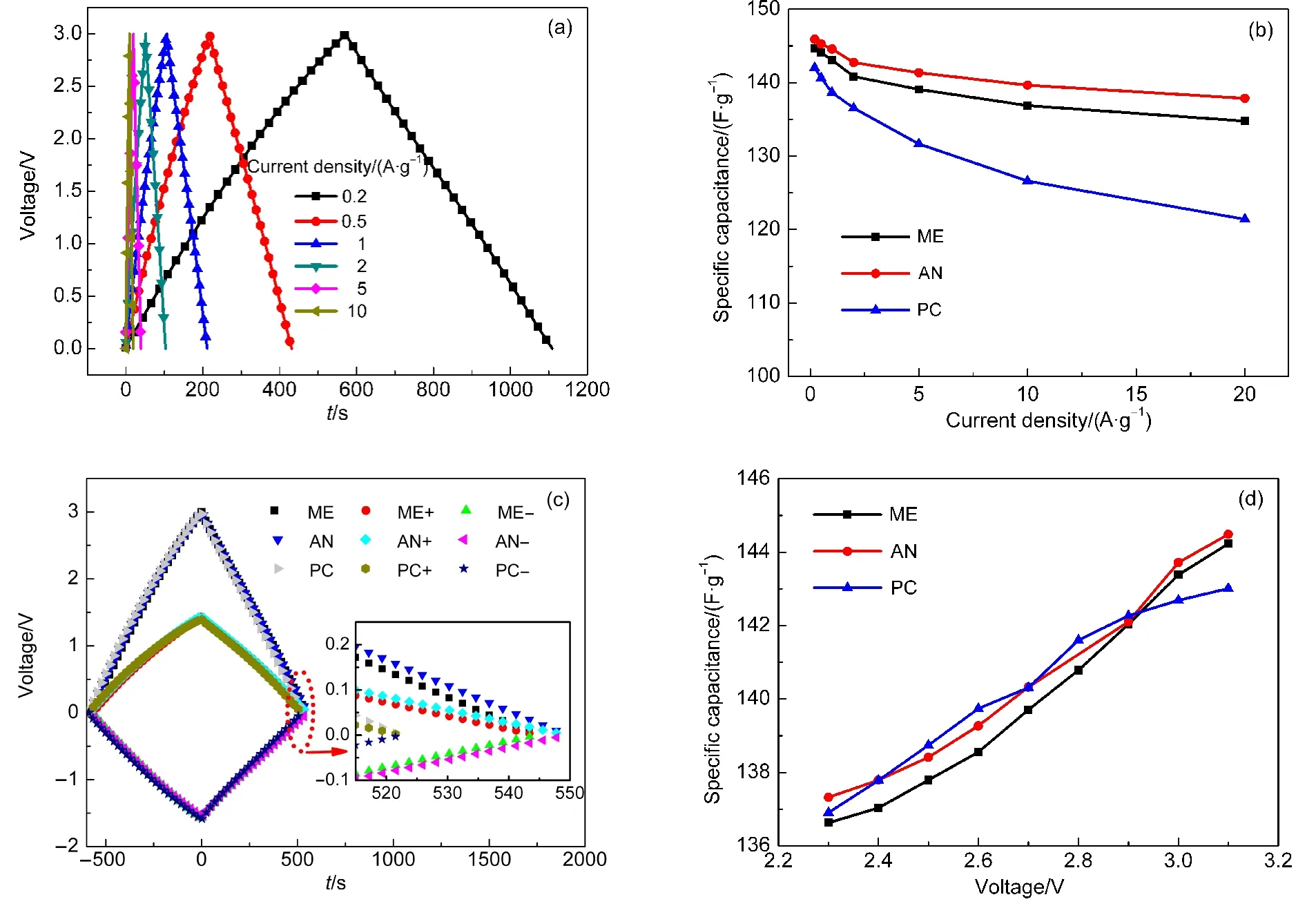
图3 超级电容器的恒流充放电曲线(a,c)和比电容曲线(b,d)Fig.3 Galvanostatic charge-discharge curves(a,c)and specific capacitance curves(b,d)of supercapacitor
图3(d)是不同截止电压下,三种超级电容器的比电容变化情况,可见,随着截止电压的提高,三种电解液的比电容均有所提高,比电容与工作电压大致为线性关系,这一现象可由CV曲线加以解释:由截止电压升高后的CV曲线可见,在升高前的截止电压范围内的电流要明显低于电压升高范围的电流,这样整个电压窗口的平均值将有所增加,即比电容有所提高.比如由图1(a)可见,3.1 V下的CV曲线较2.9 V下的CV曲线的电压升高范围为2.9−3.1 V,在这一范围内,电流较升高前的截止电压范围内(0−2.9 V)的电流明显要大,3.1 V截止电压下的CV曲线的电流均值较2.9 V截止电压下的CV曲线的均值就有所提高.同时还可见,在3 V的工作电压下,三种电解液的比电容大小顺序为AN>ME>PC,这与在3 V下CV曲线所观测到的结论是完全一致的.
3.4 漏电流和自放电测试
漏电流主要由以下三个方面引起:(1)由电极或电解质中存在的杂质引起的微电化学电池电流;(2)由电势梯度和离子浓度差引起的双电层电荷扩散电流;(3)集流体或壳体毛刺刺穿隔膜所引起的微短路电流.漏电流测试的主要作用是衡量超级电容器的绝缘性能.如图4可知,恒压充电大约前1000 s,PC的漏电流密度要高于ME和AN,但是PC的漏电流密度下降速度比ME和AN要快.大约在第3000 s左右,三种超级电容器的漏电流密度已基本稳定.在2 h时,三种超级电容器漏电流密度依次为4.45、7.43、4.41 mA·g−1,可见ME电解液的漏电流密度与PC极为相似,AN的漏电流密度要明显比ME和PC的高.

图4 不同电解液超级电容器的漏电流密度(LCD)和放大图(插图)Fig.4 Leakage current density(LCD)of supercapacitors with different electrolytes and amplified plots(inset)

图5 超级电容器比电容保持率和库仑效率与循环次数的关系Fig.5 Evolution of the specific capacitance retention and coulombic efficiency versus the number of cycles
自放电测试主要目的是为了衡量超级电容器长时间存储电荷的能力,本文自放电的测试方法是:在超级电容器漏电流测试结束后(超级电容器此时电压为3 V),将超级电容器静置12 h,然后测量超级电容器保持的电压.漏电流是引起超级电容器电压随搁置时间降低的主要原因.三种电解液下的电压分别为:2.2778、0.9623、2.4267 V,可见,AN电压损失最大,这与其有较大的漏电流相吻合.ME电解液与PC相似,但比PC稍差.
3.5 循环寿命和库仑效率
循环寿命是衡量超级电容器质量好坏的一个重要指标.随着充放电循环的增加,电解液不断分解,累积的电解液分解产物会堵塞活性炭发达的孔洞,导致极片的有效表面积减少,从而使电容量下降.29,30
图5是三种超级电容器在3 V下的循环寿命和库仑效率曲线,其充放电电流密度为1 A·g−1.超级电容器的容量随着循环次数的增加而减小,尤其是在开始1000周衰减较大,AN在3 V下的循环表现最差,PC稍微优于ME但总体差别不大.在第5000周处,ME电解液容量保持率为83.90%,PC保持率为84.63%,AN保持率为17.39%,可见AN的循环寿命远不及ME和PC.同时,由库仑曲线知,AN的效率约为98.5%,比ME和PC明显要差,ME和PC的效率约为99.8%,可见ME的库仑效率与PC相似.
4 结 论
使用一种新型有机混合电解液(三乙基甲基四氟硼酸铵/(丙烯碳酸酯+乙腈))和两种传统的传统的电解液(四乙基四氟硼酸铵/丙烯碳酸酯和四乙基四氟硼酸铵/乙腈),分别制作成活性炭基软包装超级电容器,并在3 V下对三种超级电容器进行了综合比较,经比较发现,新型的混合电解液明显吸收了AN和PC各自的优点,电导率高达40 mS·cm−1,且比电容大、漏电流小、循环寿命好、库仑效率高,综合性能优异.
致谢: 感谢北京化学试剂研究所提供本文所使用的新型有机电解液.
(1)Yu,P.;Zhang,X.;Chen,Y.;Ma,Y.;Qi,Z.Mater.Chem.Phys.2009,118,303.doi:10.1016/j.matchemphys.2009.07.057
(2) Zhang,D.;Zhang,X.;Chen,Y.;Wang,C.;Ma,Y.;Dong,H.;Jiang,L.;Meng,Q.;Hu,W.Phys.Chem.Chem.Phys.2012,14,10899.doi:10.1039/c2cp41051f
(3)Ammam,M.;Fransaer,J.J.Electrochem.Soc.2011,158,A14.
(4) Pandolfo,A.G.;Hollenkamp,A.F.J.Power Sources 2006,157,11.doi:10.1016/j.jpowsour.2006.02.065
(5) Sun,X.;Zhang,X.;Zhang,H.;Zhang,D.;Ma,Y.J.Solid State Electrochem.2012,16,2597.doi:10.1007/s10008-012-1678-7
(6) Kurig,H.;Janes,A.;Lust,E.J.Electrochem.Soc.2010,157,A272.
(7) Zhong,H.X.;Zhao,C.B.;Luo,H.;Zhang,L.Z.Acta Phys.-Chim.Sin.2012,28,2641.[仲皓想,赵春宝,骆 浩,张灵志.物理化学学报,2012,28,2641.]doi:10.3866/PKU.WHXB201207181
(8) Lu,W.;Hartman,R.;Qu,L.T.;Dai,L.M.J.Phys.Chem.Lett.2011,2,655.doi:10.1021/jz200104n
(9) Kim,B.;Chung,H.;Kim,W.Nanotechnology 2012,23.
(10) Sun,X.Z.;Zhang,X.;Zhang,D.C.;Ma,Y.W.Acta Phys.-Chim.Sin.2012,28,367.[孙现众,张 熊,张大成,马衍伟.物理化学学报,2012,28,367.]doi:10.3866/PKU.WHXB201112131
(11) Liu,P.;Verbrugge,M.;Soukiazian,S.J.Power Sources 2006,156,712.doi:10.1016/j.jpowsour.2005.05.055
(12) Portet,C.;Taberna,P.L.;Simon,P.;Flahaut,E.J.Power Sources 2005,139,371.doi:10.1016/j.jpowsour.2004.07.015
(13) Sillars,F.B.;Fletcher,S.I.;Mirzaeian,M.;Hall,P.J.Phys.Chem.Chem.Phys.2012,14,6094.doi:10.1039/c2cp40089h
(14) Lewandowski,A.;Olejniczak,A.;Galinski,M.;Stepniak,I.J.Power Sources 2010,195,5814.doi:10.1016/j.jpowsour.2010.03.082
(15) Pandey,G.P.;Kumar,Y.;Hashmi,S.A.Indian J.Chem.Sect.A-Inorg.Bio-Inorg.Phys.Theor.Anal.Chem.2010,49,743.
(16) Kurig,H.;Vestli,M.;Tonurist,K.;Janes,A.;Lust,E.J.Electrochem.Soc.2012,159,A944.
(17) Ruiz,V.;Huynh,T.;Sivakkumar,S.R.;Pandolfo,A.G.RSC Adv.2012,2,5591.doi:10.1039/c2ra20177a
(18) Balducci,A.;Dugas,R.;Taberna,P.L.;Simon,P.;Plee,D.;Mastragostino,M.;Passerini,S.J.Power Sources 2007,165,922.doi:10.1016/j.jpowsour.2006.12.048
(19)Tyunina,E.Y.;Afanasiev,V.N.;Chekunova,M.D.Journal of Chemical&Engineering Data 2011,56,3222.doi:10.1021/je200309v
(20) Moumouzlas,G.;Panopoulos,D.K.;Ritzoulis,G.Journal of Chemical and Engineering Data 1991,36,20.doi:10.1021/je00001a006
(21) Borenstien,A.;Noked,M.;Okashy,S.;Aurbach,D.J.Electrochem.Soc.2013,160,A1282.
(22)Wang,G.X.;Shao,Z.P.;Yu,Z.L.Nanotechnology 2007,18,205705.
(23)Chen,H.;Wang,F.;Tong,S.S.;Guo,S.L.;Pan,X.M.Appl.Surf.Sci.2012,258,6097.doi:10.1016/j.apsusc.2012.03.009
(24)Liu,X.M.;Zhang,R.;Zhan,L.;Long,D.H.;Qiao,W.M.;Yang,J.H.;Ling,L.C.New Carbon Mater.2007,22,153.doi:10.1016/S1872-5805(07)60015-8
(25) Ferg,E.;Rossouw,C.;Loyson,P.J.Power Sources 2013,226,299.doi:10.1016/j.jpowsour.2012.10.087
(26) Kotz,R.;Carlen,M.Electrochim.Acta 2000,45,2483.doi:10.1016/S0013-4686(00)00354-6
(27) Lust,E.;Jänes,A.;Arulepp,M.J.Electroanal.Chem.2004,562,33.doi:10.1016/j.jelechem.2003.07.034
(28) Arulepp,M.;Permann,L.;Leis,J.;Perkson,A.;Rumma,K.;Jänes,A.;Lust,E.J.Power Sources 2004,133,320.doi:10.1016/j.jpowsour.2004.03.026
(29) Ruch,P.W.;Cericola,D.;Foelske-Schmitz,A.;Kötz,R.;Wokaun,A.Electrochim.Acta 2010,55,4412.doi:10.1016/j.electacta.2010.02.064
(30) Ruch,P.W.;Cericola,D.;Foelske,A.;Kötz,R.;Wokaun,A.Electrochim.Acta 2010,55,2352.doi:10.1016/j.electacta.2009.11.098
