分级结构ZnO六棱柱:制备及对5-取代-1氢四氮唑催化性能
2013-09-15郎雷鸣柳闽生
郎雷鸣 柳闽生
(南京晓庄学院生物化工与环境工程学院,南京 211171)
由于分级结构的纳米材料具有独特的物理、化学和光电等性能长期以来一直受到关注,而通过自组装的方法将低维纳米材料组装成具有一定形状的分级结构,是当前研究的热点领域[1-6]。这些分级结构已经在催化、药物传输、光学材料和电池材料等领域表现出了巨大的应用前景[7-11]。随着纳米科学的快速发展,许多不同物质的分级结构纳米材料已经被合成出来。如由纳米片组装成的Ni(OH)2管、Co3O4空心球以及由纳米棒自组装而成的CePO4空心球及海胆状BiPO4等材料[12-15],由纳米棒组装成的V2O5空心球[16],纳米线组装成的海胆状MnO2球以及通过自组装方法合成的纳米棒组成的CuO空心微球、海胆状Bi2S3以及由纳米线组成的CeVO4空心球等材料[17-19]。
在众多的纳米材料中,纳米ZnO是一种新型的直接宽带隙多功能半导体材料,除了具有优异的光学、电学性能外,同时集压电性能,储氢性能,热释电性能,场致发射效应,气敏性,光催化,传输特性等多种性能于一身,使其能够应用在多种不同领域[20-23]。目前关于ZnO纳米材料的研究与日俱增,相继合成出多种形貌的ZnO纳米材料,如通过气相法获得了纳米棒、纳米线、纳米同轴电缆、纳米环、钉状、梳状、螺旋状以及多种分级结构等[24-30],通过化学液相法或电化学方法获得了花状、针状、哑铃状、塔状、管状以及阵列等结构的ZnO[31-35],而通过简单有效的液相法在较低温度下自组装成具有多级生长的六棱柱型ZnO目前并不多见,仅有为数不多的报导关于哑铃型分级结构的ZnO的合成[35-38],但其合成方法为水热法,需要较高的温度和压力,条件要求较高。本工作主要通过简单有效的液相法在较低温度下通过自组装获得了具有分级结构的ZnO六棱柱,并对其形成过程进行了探讨,该方法简单易操作,便于大规模合成,同时对ZnO六棱柱进行了四氮唑催化反应研究,显示了好的催化活性。
1 实验部分
1.1 试 剂
醋酸锌(Zn(Ac)2·2H2O)、SDS(十二烷基磺酸钠)、CTAB(十六烷基三甲基溴化铵)、PEG20000(聚乙二醇20000)、PEG400(聚乙二醇 400)、F127(PEO-PPO-PEO,三嵌段共聚物)、乙二醇、蒸馏水、无水乙醇、六亚甲基四胺,叠氮钠(NaN3),N,N-二甲基甲酰胺(DMF),苯甲腈,化学试剂均为分析纯,使用前未经进一步提纯。
1.2 分级结构ZnO的制备
准确称取 2 mmol Zn(Ac)2·2H2O、2 mmol六亚甲基四胺、0.2 g CTAB于100 mL圆底烧瓶中,加入30 mL蒸馏水作为溶剂,将样品置于超声仪中超声,直至溶液完全溶解至透明状态,再将溶液置于85℃的油浴下磁力搅拌反应2 h后取出,以5 000 r·min-1的转速离心分离产品,并用无水乙醇洗涤2~3次,将所得产品放入烘箱干燥,得到产品供各项表征。
1.3 表 征
用X射线衍射仪(XRD-6000日本岛津公司,Cu Kα1射线,波长 0.154 06 nm,工作电压 40 kV,电流 30 mA, 扫描速度为 6°·min-1, 扫描范围 20°~80°),透射电子显微镜 (TEM)、高分辨透射电镜(HRTEM)和扫描电子显微镜(SEM,JEOL S-4800,加速电压15 kV)等对产物的形貌、相结构和微观结构进行表征。TEM的型号为Japan JEOL JEM-200CX,加速电压为200 kV,并装有快速傅立叶变换(FFT)和能谱 (EDS)测量附件。高分辨透射电子显微镜(HRTEM)为Philips TECNAI F-30 FEG,加速电压为300 kV。样品的表面元素价态通过X射线光电子能谱(XPS,VGESCALAB MKII)进行分析,使用单色化Al Kα 射线源(hν=1 486.6 eV),加速电压和工作电流为 12.5 kV 和 20.0 mA。
1.4 5-取代-1氢四氮唑的合成
5-取代-1氢四氮唑的合成步骤如下:以苯甲腈为反应底物为例,将 0.1 g ZnO 六棱柱,0.257 g 苯甲腈(2.5 mmol)、0.35 g 叠氮钠(5.4 mmol)和 5 mL 溶剂(DMF)加入到25 mL圆底烧瓶中,放入油浴,烧瓶口上端接上冷凝管,冷凝管末端与干燥管相连,磁力搅拌下升温至120℃,在此温度下反应12~36 h,其间用薄层色谱跟踪反应进程,反应进行36 h后,停止搅拌和加热,待体系冷却至室温后,离心分离催化剂,用5 mL乙酸乙酯洗涤催化剂3次,向分离液中加入乙酸乙酯(15 mL)和 6 mol·L-1HCl(20 mL),搅拌5 min,分出有机层,再用乙酸乙酯萃取水层,每次用量20 mL,萃取5次,合并有机相,用20 mL水洗两次后,旋转蒸发除去有机溶剂,得到白色固体,经50℃真空干燥8 h后,称取质量,计算收率(以苯甲腈为基准)。再经过柱色谱提纯后,所有四氮唑产品进行了1H NMR,IR 表征,1H NMR(300 MHz,CDCl3):δ=8.00(m,2H),7.59(m,3H),MS(70 eV):m/z(%)=146.1(M+,6.87%),118.1(M-N2,100.0%),103.1(M-HN3,10.3%),91.1(M-HCN3,30.35%),77.1(M-HCN4,14.93%),63.0(M-H3C2N4,9.72%),and 51.0(M-H3C3N4,5.73%)。与文献数据对照,确定其结构。适当更换反应底物、改变催化剂种类及用量和改变反应温度以及反应时间,得到了一系列结果。
2 结果与讨论
2.1 XRD、XPS及BET测试结果与讨论
图1a为分级结构ZnO的XRD图,从图中可以看出ZnO的(100)、(002)、(101)3个晶面对应的衍射峰强度较大,为ZnO特征晶面衍射峰。图中每个峰的位置都与标准卡片(PDF No.36-1451)六方相ZnO的图非常吻合,没有其他杂质峰出现,说明所制备的产品为纯相的ZnO。图1b,1c分别为ZnO中锌元素和氧元素的XPS谱图,在束缚能为531.2 eV处出现的峰对应于O1s(图1b),而在1 022.0 eV和1 045.1 eV处的较强峰是则归属于Zn的2p1/2和2p3/2。图1d是六棱柱ZnO氮气吸附脱附曲线图,图中曲线属于Ⅳ型回滞曲线,比表面积(SBET)为23 m2·g-1。
2.2 SEM、TEM和HRTEM测试结果与讨论
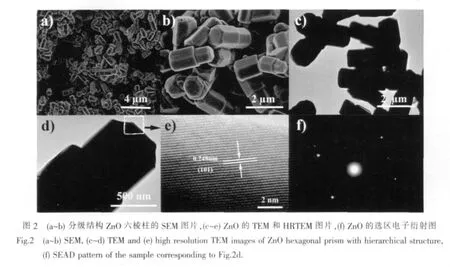
图2a和2b分别为制备的分级结构的ZnO低倍下大面积和局部放大的SEM图片,从图中可以清楚的看到所制备的ZnO分布均匀,尺寸非常均一,平均直径在1μm,长度2μm,呈现六棱柱形的分级结构 (图2b)。图2c和2d是大面积的和单个ZnO的TEM图片,从图2c中可以清楚的看出分级结构中2个六棱柱之间有明显的截面,对单个的ZnO进行放大,其分级结构清晰可见(图2d),并且从分级结构的顶部可以观察到六棱柱有进一步向外生长的趋势。图2e为图2d中ZnO顶端的高分辨电镜照片,图中显示很规整的晶格条纹,晶面间距为0.248 nm,对应于 ZnO 的(101)晶面(图 2e),选区电子衍射图中出现的亮点证明所合成的产品为单晶结构的 ZnO(图 2f)。
2.3 不同表面活性剂、碱源对产物形貌的影响
为了考察不同表面活性剂对产品形貌的影响。我 们 使 用 了 多 种 表 面 活 性 如 CTAB,SDS,F127,PEG20000,PEG400等。图3a是在无表面活性剂时所制备的ZnO的SEM图片,从图中可以看到所制备的产品多为杂乱无章的盘状、柱状结构,没有很规整的分级结构出现,当使用F127作为表面活性剂时获得部分分级结构的ZnO(图3b),但长短不一,尺寸不均,其中有部分多级生长的ZnO出现。表面活性剂如换成PEG20000和PEG400,可以看出有少量的分级结构出现(图3c,3d),大部分为六棱短柱状结构,此外还存有部分杂乱无章的小粒子。图3e则是使用SDS为表面活性剂所制备的ZnO扫描图片,图中显示产品为无规则的片状结构。通过对比不同表面活性剂所合成的不同形貌ZnO的SEM图片可知CTAB是制备具有分级结构ZnO六棱柱最合适的表面活性剂。
为了研究不同碱对产品形貌的影响,在表面活性剂同为CTAB的情况下,我们更换了不同的碱源。图5a是使用尿素作为碱源所得碱式碳酸锌的SEM图,可以看出制备的产品是比较规整的短圆柱形结构,表面有部分孔洞,有些短圆柱则呈现完全的空心结构。可能由于在反应过程中尿素分解产生过量的NH3和CO2形成气泡,而气泡在高温下涌出导致空心结构的形成。碱式碳酸锌受热后所得ZnO的形貌依然保持了短圆柱型结构(图5b),当碱换成NaOH时,所得产品为杂乱无章、无规则的ZnO粒子 (图5c)。图5d则是使用氨水作为碱源时产品的SEM图,图中显示制备的产品为纳米棒组成的花状结构。由此可见,在相同条件下不同碱源的使用可对产品形貌产生重要影响,而采用六亚甲基四胺作为碱源,可以获得规整的分级结构的ZnO六棱柱(图 2a,2b)。

2.4 分级结构ZnO六棱柱形成过程分析
在合成纳米材料过程中,表面活性剂的使用对产品形貌有很大的影响,为了研究在表面活性剂CTAB辅助下分级结构ZnO六棱柱的形成过程,通过调控CTAB的用量获得了一系列不同形貌的ZnO六棱柱,并根据形貌的变化初步推断分级结构形成原因。当使用0.1 g CTAB时,获得的大部分是六棱短柱ZnO,很少有分级结构的ZnO出现 (图5a),而CTAB的用量增大到0.2 g时,获得了很规整的二级生长结构的ZnO(图5b),继续增大CTAB的用量到0.3 g时,大部分为短的六棱柱,也有少量分级结构出现(图 5c),而 CTAB 用量达到 0.5 g时,几乎全为尺寸较小的六棱短柱 (图5d)。由此可见CTAB的浓度对分级结构ZnO六棱柱的形成有重要影响。
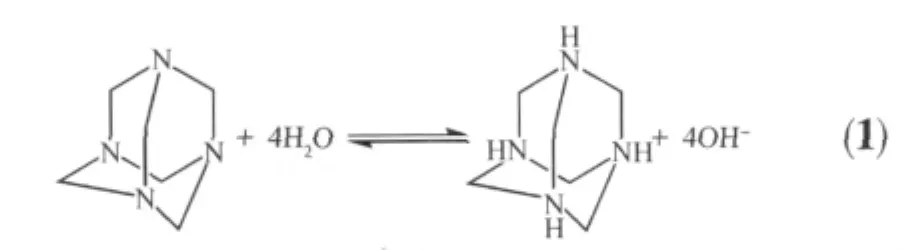
以上实验可以推断出,在该体系中,当温度达到90℃时六亚甲基四胺开始分解生成OH-(方程1),溶液中 Zn2+与过量 OH-的结合生成,当浓度超过临界浓度时则生成ZnO微晶,此时如果表面活性剂的浓度较低则覆盖在ZnO表面的CTAB较少,ZnO微晶受表面活性剂的影响较小,形成较大的六棱柱,而当CTAB浓度较高时,覆盖在ZnO表面的CTAB较多,阻碍了ZnO微晶进一步向外生长,所得产品的尺寸就相对较小,六棱柱的形貌也欠规整(图5d),当覆盖在ZnO表面的CTAB的浓度比较适中,此时在ZnO六棱柱的侧面和顶端都有可能进行附着生长,但ZnO六棱柱顶端的表面能较高,活性位点更多,更有利于的附着,进而形成二级生长的分级结构。产品形貌随着反应时间的变化,更能清楚的说明这种变化趋势。图6a~d是在CTAB的用量为0.2 g时分别反应30 min,1 h,4 h,6 h所得产品的SEM图片。图6a为反应30 min时ZnO的SEM图片,图中显示有分级结构和短的六棱柱ZnO出现,但大部分为六棱短柱,说明此时正处于ZnO顶端二级生长的初始阶段,当反应时间延长至1 h,获得的产品大部分为分级结构的ZnO(图6b),而反应2 h后,制备的ZnO全为规整的二级生长的六棱柱结构(图2a),此时已达二级生长的最佳时间,继续延长反应时间至4 h(图6c)和6 h(图6d),可以获得两端生长的分级结构的ZnO,说明延长反应时间有利于ZnO六棱柱在活性较高的顶端继续向外生长,由此也很好的证明了我们所提出的分级结构ZnO六棱柱的形成机理。

2.5 5-取代-1氢四氮唑催化反应
四氮唑是一类含氮的杂环、具有广泛用途的物质,在许多方面有实际的和潜在的用途[39-41]。我们利用分级结构氧化锌六棱柱作为催化剂来制备四氮唑并且取得了良好的催化效果。表1是对各种反应条件(包括反应时间,温度,溶剂,有无催化剂)进行实验后所得结果的汇总,从此表中可以看出,反应进行 12 h,产率为 50%(表 1,1),反应 24 h 后,产率提高到69%(表1,2),当反应时间为36 h,产率则达到85%(表1,3),如果再继续延长反应时间,则产率不会再有所增加 (表1,4),所以最佳的反应时间为36 h。我们对反应温度进行摸索,发现110℃时产率比较低(表1,5),而130°C时产率提高也不是很多(表1,6),所以最佳反应温度为120℃。考虑溶剂对反应的影响,我们尝试了多种溶剂,如DMSO,水,THF,发现这些溶剂都不太适合该反应,尤其是水和THF,产率很低,所以最佳溶剂为 DMF(表 1,7~9)。在以上条件确定的情况下,为了对比,不用任何催化剂进行反应,结果发现没有任何产品生成,说明不用催化剂不能催化该反应进行(表1,10)。此外,对不同条件下合成的ZnO进行催化性能研究发现,在高温下退火后的ZnO六棱柱催化活性稍高于原始产品(表1,11),以尿素合成的碱式碳酸锌经过煅烧后得到的ZnO催化性能要略好于ZnO六棱柱 (表1,12),而其他几种形貌的ZnO与六棱柱ZnO催化效果相当(表1,13~14),以上结果表明产品的形貌和表面结构对催化性能有一定的影响。
在最优化条件下以分级结构ZnO六棱柱为催化剂对不同种类的腈进行催化活性研究,如表2所示,我们选择了8种不同的反应底物,大部分产率都达到85%以上,尤其是4-氰基吡啶显示了非常高的催化效果,收率达92%(表2,8),而苯乙腈由于基团腈没有直接与苯环相连显示了较差的活性 (表2,7)。基团所在的位置不同也对产率有一定的影响,含有间位和对位的基团如3-氯,4-氯,4-硝基等都显示了比较好活性(表2,3~5),而基团在邻位的底物如 2-氯苯甲腈(表 2,2),收率较低,只有 75%,这主要是由于在邻位的基团阻碍了唑的形成。另外苯环上连有供电子基团的甲基也有很高的收率(表2,6)。
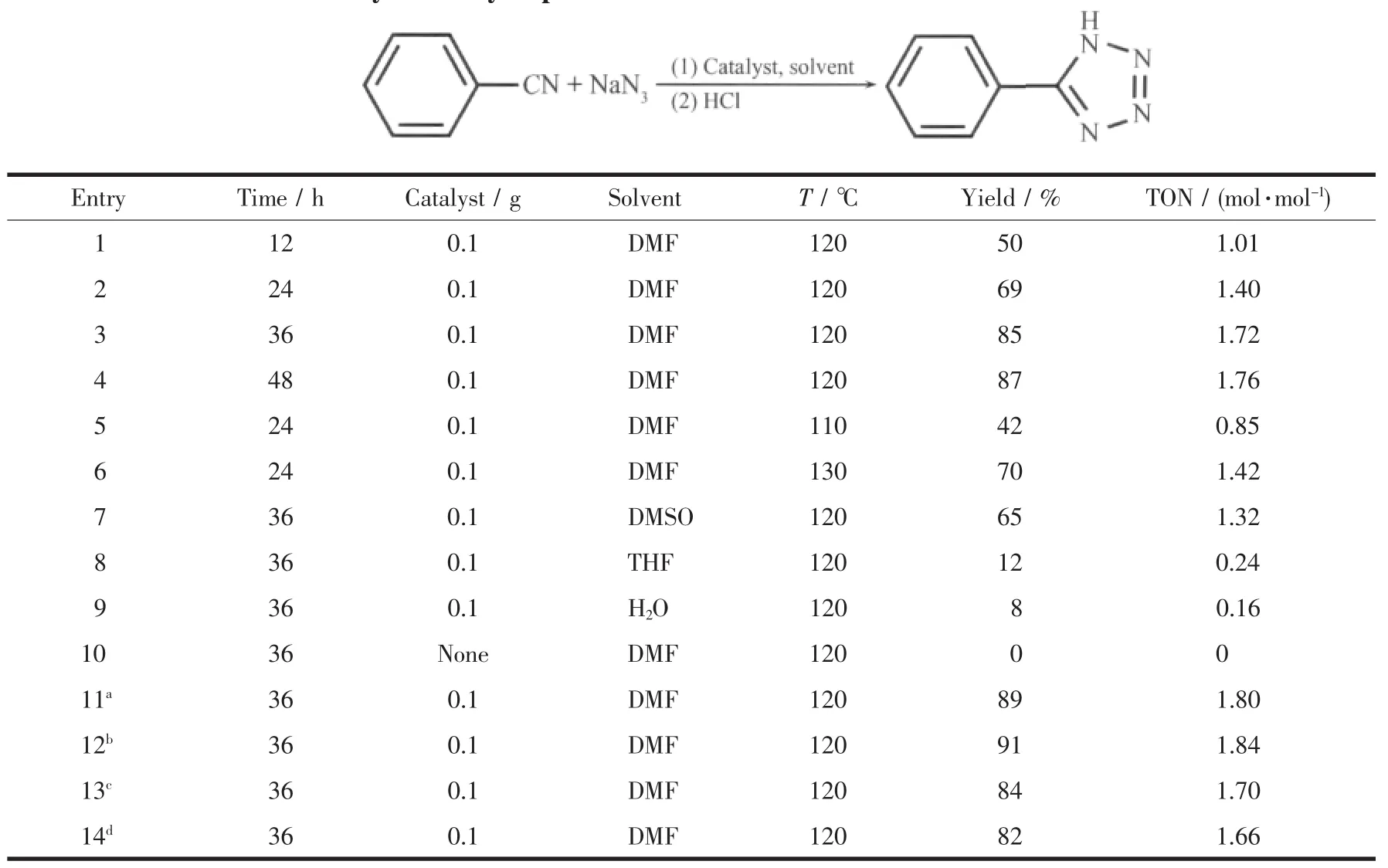
表1 不同反应条件下ZnO催化性能汇总表Table 1 Summary of catalytic performance of ZnO under different reaction conditions
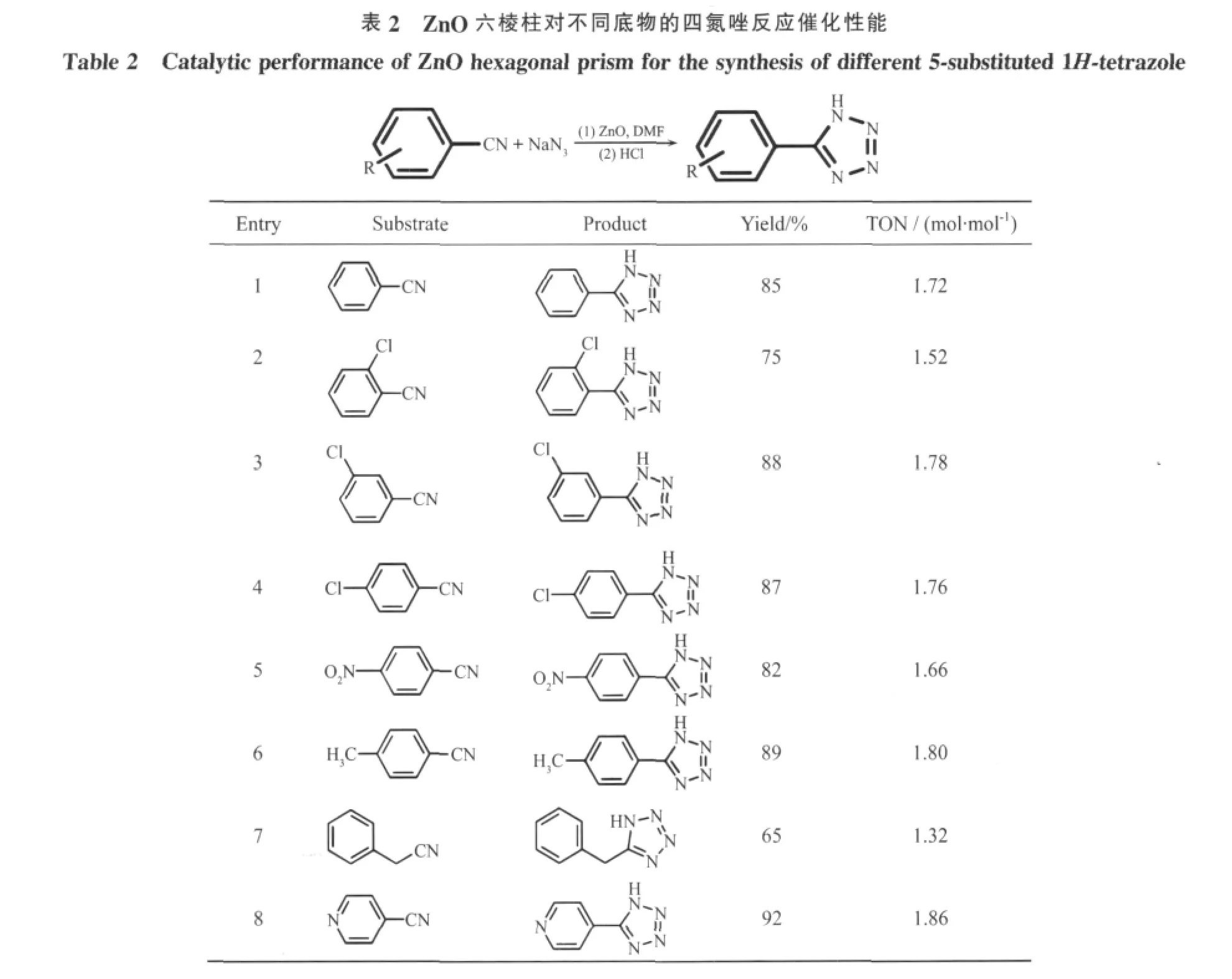
?
3 结 论
本论文主要以简便有效地液相法在表面活性剂CTAB的辅助下制备了具有分级结构的ZnO六棱柱,通过运用 XRD,XPS,SEM,TEM,HRTEM 等手段对所合成的样品进行表征和分析,详细考察了不同表面活性剂、不同碱源对产品形貌的影响,结果表明CTAB和六亚甲基四胺为最佳的表面活性剂和碱源。此外,本文系统研究了CTAB的浓度以及反应时间对产品形貌的影响,初步推断出分级结构ZnO六棱柱的形成机理,为实现分级结构ZnO纳米材料的可控合成提供了一定实验和理论依据。同时以ZnO六棱柱为催化剂通过[3+2]环加成反应催化合成5-取代-1-氢四氮唑,系统考察了各种反应条件对该反应的影响,筛选出最佳条件,并用苯甲腈衍生物进行了[3+2]环加成反应研究,显示了很好的催化效果,产率大多在85%以上。
[1]Park S,Lim JH,Chung SW,et al.Science,2004,303:348-351
[2]Cao A M,Hu JS,Liang H P,et al.Angew.Chem.Int.Ed.,2005,44:4391-4395
[3]Liu B,Zeng H C.J.Am.Chem.Soc.,2004,126:16744-16746
[4]Mo M S,Yu JC,Zhang L Z,et al.Adv.Mater.,2005,17:756-760
[5]Xu L,Ding Y S,Chen C H,et al.Chem.Mater.,2008,20:308-316[6]Cheng Y,Wang Y S,Jia C,et al.J.Phys.Chem.B,2006,110:24399-24402
[7]Gao PX,Wang Z L.J.Am.Chem.Soc.,2003,125:11299-11305
[8]Hu J,Bando Y,Zhan J,et al.Adv.Mater.,2005,17:1964-1969
[9]Zhou J,Ding Y,Deng SZ,et al.Adv.Mater.,2005,17:2107-2110
[10]Lou X W,Deng D,Lee J Y,et al.Adv.Mater.,2008,20:258-262
[11]Zhang L,Wang W,Chen Z,et al.J.Mater.Chem.,2007,17:2526-2532
[12]Tao F,Guan M,Zhou Y,et al.Crystal Growth.Des.,2008,8:2157-2162
[13]Guan M,Sun J,Tao F,et al.Crystal Growth Des.,2008,8:2694-2697
[14]Guan M,Sun J,Han M,et al.Nanotechnology,2007,18:415602-415607
[15]Tao F,Gao C,Wen Z,et al.J.Solid State Chem.,2009,182:1055-1060
[16]Yuan J K,Li W N,Gomez S,et al.J.Am.Chem.Soc.,2005,127:14184-14185
[17]Liu B,Zeng H C.J.Am.Chem.Soc.,2004,126:8124-8125
[18]Shen G,Chen D,Tang K,et al.Chem.Phys.Lett.,2003,370:334-337
[19]Luo F,Jia CJ,Song W,et al.Crystal Growth.Des.,2005,5:137-142
[20]Jin Z C,Hamberg I,Granqvist C G.J.Appl.Phys.,1988,64:5117-5131
[21]Pearton S J,Abernathy C R,Overberg M E,et al.J.Appl.Phys.,2003,93:1-13
[22]Pearton SJ,Norton D P,Ip K,et al.Mater.Sci.,2005,50:293-340
[23]Macdonald A H,Schiffer P,Samarth N.Nat.Mater.,2005,4:195-202
[24]Tian Z R,Voigt JA,Liu J,et al.J.Am.Chem.Soc.,2002,124:12954-12955
[25]Hu JQ,Li Q,Meng X M,et al.Chem.Mater.,2003,15:305-308
[26]Bekermann D,Gasparotto A,Barreca D,et al.Crystal Growth Des.,2010,10:2011-2018
[27]Lao J Y,Huang J Y,Wang D Z,et al.Nano Lett.,2003,3:235-238
[28]Kong X Y,Wang Z L.Nano Lett.,2003,3:1625-1631
[29]Zhang H,Yang D,Ji Y,et al.J.Phys.Chem.B,2004,108:3955-3958
[30]Lao JY,Wen JG,Ren Z F.Nano Lett.,2002,2:1287-1291
[31]Vayssieres L,Keis K,Hagfeldt A,et al.Chem.Mater.,2001,13:4395-4398
[32]Hu P,Zhang X,Han N,et al.Crystal Growth Des.,2011,11:1520-1526
[33]Zhang J,Wang S,Xu M,et al.Crystal Growth Des.,2009,9:3532-3537
[34]Jung S,Oh E,Lee K,et al.Crystal Growth Des.,2008,8:265-269
[35]Ayudhya S K N,Tonto P,Mekasuwandumrong O,et al.Crystal Growth Des.,2006,6:2446-2450
[36]Wang B G,Shi E W,Zhong W Z.Cryst.Res.Technol.,1998,33:937-940
[37]Zhang H,Yang D,Li D,et al.Crystal Growth Des.,2004,5:547-550
[38]Yu Q,Yu C,Yang H,et al.Inorg.Chem.,2007,46:6204-6210
[39]Xiong R G,Xue X,Zhao H.Angew.Chem.Int.Ed.,2001,41:3800-3803
[40]Ye Q,Song Y M,Wang G X.J.Am.Chem.Soc.,2006,128:6554-6555
[41]Huisgen R,Sarer J,H.Sturm J,et al.Chem.Ber.,1960,93:2106-2124
