一种改进的赖氨酸浓度测定方法
2013-09-13姜安娜曹名锋李庆刚郑平杨洪江孙际宾
姜安娜 曹名锋 李庆刚 郑平 杨洪江 孙际宾
赖氨酸是世界上仅次于谷氨酸的第二大氨基酸品种,目前主要应用于饲料添加剂[1-3]、食品添加剂和医药中间体[4-8]。随着科技的发展,赖氨酸品种已经发展出赖氨酸盐酸盐、蛋白赖氨酸和液体赖氨酸等[9,10]。截止到2010年底,世界市场每年的赖氨酸需求量达140万吨,按照工业赖氨酸计算,其直接市值达200亿元,间接市场难以估量[11]。
目前常用的赖氨酸检测方法主要有酶法、高效液相色谱(HPLC)和化学法(茚三酮)法。酶法主要是通过赖氨酸在特异的氧化酶催化下发生氧化还原反应,其电子产物在生物传感器电极表面形成电流从而进行测定,检测准确度和应用性受酶活力和酶价格的限制;HPLC方法采用2,4-二硝基氟苯(DNFB)、1-氟-2,4-二硝基苯基 -5-L-丙氨酰胺(FDAA)等衍生化试剂,虽然能够精确区分和测定D-/L-型赖基酸的含量,但对仪器精密度和操作人员要求极高,并且易受样品中杂质成分的干扰,不适合工业发酵液中赖氨酸的检测;化学法主要指茚三酮方法,基本原理为:氨基酸与水合茚三酮共同加热后可发生氧化脱氨反应,生成NH3与酮酸;加热过程中酮酸裂解,放出CO2,自身变为少一个碳的醛,水合茚三酮变为还原型茚三酮。NH3与水合茚三酮及还原茚三酮脱水缩合,生成蓝紫色化合物[12]。传统的茚三酮检测方法无法排除杂质氨基酸的干扰,且反应过程中易受pH和温度变化的影响,造成测定结果准确性降低[13,14]。本研究在传统的茚三酮试剂中添加了铁离子(Fe3+),通过铁离子对脯氨酸、鸟氨酸、甘氨酸、精氨酸、组氨酸与茚三酮反应的抑制作用,从而提高茚三酮溶液与赖氨酸反应的特异性[15]。此外,由于赖氨酸发酵过程中产生其他副产物会影响发酵液的pH值,对反应的成色速度和产物的颜色造成影响,进而削弱了分子内基团反应的灵敏性,所以试验中拟设计不同的缓冲液反应体系,旨在提高茚三酮法检测赖氨酸的特异性,并实现了对赖氨酸发酵液稳定和准确的检测。本方法不需要昂贵的设备,方法简单,试剂便宜,数据精确,适合实验室检测和工业应用。
1 材料与方法
1.1 材料
1.1.1 化学试剂 21种氨基酸,即甘氨酸(Gly)、丙氨酸(Ala)、缬氨酸(Val)、亮氨酸(Leu)、异亮氨酸(Ile)、苯丙氨酸(Phe)、脯氨酸(Pro)、色氨酸(Trp)、丝氨酸(Ser)、酪氨酸(Tyr)、半胱氨酸(Cys)、蛋氨酸(Met)、天冬酰胺(Asn)、谷氨酰胺(Gln)、苏氨酸(Thr)、天冬氨酸(Asp)、谷氨酸(Glu)、赖氨酸(Lys)、精氨酸(Arg)、组氨酸(His)和鸟氨酸(Ornithine)购自上海生工生物工程有限公司;分析纯的茚三酮、氯化铁、甲基溶纤剂、二甲基亚砜和正己烷购自国药集团化学试剂有限公司;色谱纯乙酸钠、异硫氰酸苯酯和三乙胺购自Sigma公司;乙腈由Merck公司提供。
1.1.2 仪器和设备 酶标仪为美国Molecular Devices公司产品,恒温加热器购自上海一恒科技有限公司,电子天平(0.000 1 g)购自上海梅特勒-托利多仪器有限公司,高效液相色谱仪购自日本岛津公司。
1.2 方法
1.2.1 茚三酮-铁溶液与氨基酸溶液反应[16]首先配制200mmol/L的氨基酸溶液(天冬氨酸、谷氨酸、苯丙氨酸、异亮氨酸、亮氨酸、蛋氨酸、天冬酰胺、色氨酸、酪氨酸溶于0.1mol/L的盐酸,然后用相同体积的0.1mol/L的氢氧化钠中和,添加去离子水至终浓度200mmol/L;谷氨酰胺由于在酸碱条件下均不稳定,故配成饱和溶液,现配现用;其他氨基酸溶于去离子水)。取20μL氨基酸溶液加入到0.66mL溶液I[373mL甲基溶纤剂与30mL 50%(W/W)
的氯化铁溶液混合加入600mL缓冲液]和0.37mL溶液II(1 g茚三酮溶于100mL缓冲液)中混合均匀,100℃恒温加热40min,快速用自来水冷却至室温,加入4mL二甲基亚砜和6mL去离子水,取200μL样品在480nm波长下(常温)测定吸光值。
对显色反应的最佳反应时间、最佳检测波长进行了摸索,以确定最佳反应条件。另外,还用不同pH值的酸性氯化钾溶液取代缓冲液来确定溶液显色反应的最佳pH值以及更换不同的缓冲液确定最佳缓冲体系。
1.2.2 高效液相色谱法(HPLC)检测赖氨酸生产菌发酵液中赖氨酸的浓度[17]异硫氰酸苯酯柱前衍生-反相高效液相色谱法测定赖氨酸:将赖氨酸生产菌的发酵液分别稀释20、30和40倍,取出0.4mL置于离心管中,加入1mol/L三乙胺-乙腈溶液0.2mL,异硫氰酸苯酯乙腈溶液0.2mL,漩涡混合器振荡1min,37℃水浴1h,加入正己烷0.7mL漩涡混合器振荡1min,静置60min。用注射器吸取下层溶液,经0.45μm滤膜过滤后置于色谱自动进样系统,进行洗脱和谱图分析。
2 结果
2.1 茚三酮法最适检测条件的摸索
2.1.1 最佳检测波长和最佳pH的确定 200mmol/L赖氨酸与pH为0.8、1.6、2.4、3.2和4.0的茚三酮-铁混合液反应,等体积去离子水取代200mmol/L赖氨酸加入反应体系作为对照,通过酶标仪快速读取吸光值,检测结果见图1。对不同pH值的反应混合液进行了波谱扫描发现,最佳扫描波长为480nm。当pH>2.4时,反应溶液中出现了沉淀,所以试验选择在pH<2.4(即pH为2.2)的环境中进行。
2.1.2 最佳缓冲液的选择 将不同浓度的赖氨酸溶液(0、50、100、150、200、300、400 和 500mmol/L)与茚三酮-铁试剂在pH2.2的不同类型的缓冲液体系下进行反应,检测480nm处的吸光值。结果(图2)显示,以甘氨酸-盐酸和邻苯二甲酸-盐酸作为反应的缓冲液体系时,产物在480nm处的吸光值始终处于很小数值范围内,波形比较缓和。而在柠檬酸-氢氧化钠-盐酸和磷酸氢二钠-柠檬酸缓冲液体系中进行反应时,480nm处的吸光值较强,呈线性分布,说明柠檬酸-氢氧化钠-盐酸和磷酸氢二钠-柠檬酸缓冲液是比较理想的缓冲试剂。本试验选择pH2.2 的磷酸氢二钠-柠檬酸缓冲液进行后续的试验。
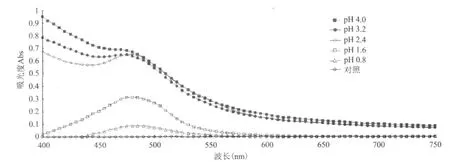
图1 不同pH茚三酮-铁试剂与200mmol/L赖氨酸反应的吸光值

图2 不同浓度的赖氨酸溶液与茚三酮-铁试剂在不同缓冲液体系下反应结果

图3 不同浓度的赖氨酸与茚三酮-铁试剂在100℃反应的变化
2.1.3 最佳反应时间的确定 选择线性关系中赖氨酸浓度最高的200mmol/L溶液,与茚三酮-铁混合溶液在100℃条件下加热,每隔5min检测反应溶液在480nm处的吸光值。结果(图3)表明,反应结束达到稳定期需要的时间基本与赖氨酸浓度成正比。当200mmol/L赖氨酸与茚三酮溶液反应,在沸水浴中加热至35min时抛物线坡度开始变缓,接近最高点,吸光值的线性关系较好,可以将100℃加热35min作为200mmol/L赖氨酸完全反应的条件;当待测试样中赖氨酸浓度低于200mmol/L时,反应时间可以适当缩短。
2.2 茚三酮法对赖氨酸检测的特异性与灵敏度
2.2.1 改进的茚三酮法对赖氨酸的特异性检测 在pH2.2 的磷酸氢二钠-柠檬酸缓冲液条件下,采用常见的21种氨基酸(200mmol/L)与茚三酮-铁试剂反应,进行350nm-650nm的波谱扫描(图4),并检测480nm处的特异性反应(表1),以反应体系中去离子水取代氨基酸为对照。从图4的波谱扫描结果可以看出,虽然茚三酮与色氨酸反应在390nm处出现最大吸收的波峰,但480nm处吸光值仅为0.02,相反在480nm处只有赖氨酸反应产物出现强吸收峰,其吸收值为0.37,远高于其他氨基酸。因此,在pH2.2的磷酸氢二钠-柠檬酸缓冲液条件下,茚三酮-铁试剂对赖氨酸的反应特异性非常强,可以完全排除其他氨基酸的干扰。
2.2.2 改进的茚三酮法对赖氨酸浓度检测范围的确定 配制浓度为0、50、100、150、200、300、400和500mmol/L的赖氨酸溶液与茚三酮-铁试剂反应,100℃加热1h,测定产物在480nm处的吸光值。从图5可以看出,数值分布趋势较明显,在赖氨酸浓度0-200mmol/L范围内其吸光值与赖氨酸浓度呈非常好的线性关系(R2=0.9924),因此茚三酮方法测定的赖氨酸适用浓度范围为0-200mmol/L。

图4 21种氨基酸(200mmol/L)与茚三酮-铁试剂反应的波谱扫描

表121种氨基酸(200mmol/L)与茚三酮-铁试剂反应结果
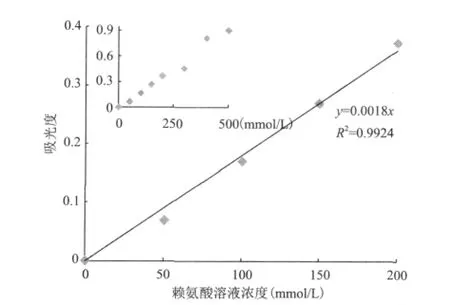
图5 不同浓度的赖氨酸与茚三酮-铁试剂反应的标准曲线

表2 茚三酮-铁和HPLC检测发酵液中赖氨酸含量比较
2.2.3 改进的茚三酮法与HPLC检测法的比较 以本研究室构建的一株高产赖氨酸的基因工程大肠杆菌经过摇瓶发酵36h后,12000r/min离心10min去除菌体,发酵液上清分别采用茚三酮-铁试剂法和反相高效液相色谱HPLC法检测产物赖氨酸的含量。表2中数据显示,茚三酮-铁试剂检测发酵液中赖氨酸含量与HPLC检测结果几乎一致,但是HPLC会出现一定的图谱重叠(谱图未给出),分析原因可能为发酵液中杂质氨基酸的干扰。茚三酮-铁法检测赖氨酸的方法可以克服这个缺点,并且在茚三酮方法简便易行,对设备和试验人员要求低,满足了工业发酵的需要。
3 讨论
在改进的茚三酮法检测赖氨酸的试验过程中,甲基溶纤剂(GDME)和二甲基亚砜(DMSO)作为有效的分散剂和促溶剂,通过加快分子间化学键的形成可以大大促进α-氨基酸和茚三酮的反应,帮助反应溶液中的混合物均匀分布,特别是在高温反应结束后加入DMSO使数据检测结果更加精确[16]。然而我们在尝试低浓度赖氨酸(<10g/L)的测定时发现,若按照上述材料方法中的4mL DMSO和6mL去离子水加入,就会导致稀释倍数过大,检测吸光度数值很小,导致误差增大,因此可以适当减小稀释倍数,比如只加1mL DMSO,在480nm下检测溶液的吸光值,也会得到很好的标准曲线,数值分布趋势较明显,在赖氨酸浓度0-6g/L范围内反应结果吸光值与赖氨酸浓度呈线性关系(R2=0.997),检测灵敏度为≤1g/L(数据未给出)。
Chinard[15]提出,在强酸性条件下,茚三酮可以与脯氨酸和鸟氨酸反应,使溶液呈现红色;Hsieh等提出,金属离子(铜、铁、锡等)能够抑制茚三酮与脯氨酸、鸟氨酸、甘氨酸、精氨酸和组氨酸的显色反应[16,18],可以一定程度上排除杂质氨基酸的干扰。刘飞飞等[19]在测定赖氨酸最佳反应条件时发现加热时间过长导致反应结果吸光值下降。此外,据报道pH的变化不仅影响到氨基酸和茚三酮反应的成色速度和产物的颜色,而且可以影响催化反应的机理,即分子内其他一些基团对茚三酮反应的灵敏性,但目前仍没有通过调节pH来降低其他氨基酸干扰的报道。因此,我们尝试在加入pH2.2的缓冲液和铁离子后,发现当茚三酮与赖氨酸反应结束后,吸光值在较长时间内保持稳定,并且反应体系的检测结果也非常灵敏。
随着工业生物技术的发展,菌种、药物等目的试样的快速筛选变得极为重要,这就需要一套有效的高通量筛选技术来实现小反应体积、高灵敏度和短反应时间的测定。本研究中介绍的方法,当温度设置为100℃时,容易造成溶剂的挥发,使得反应体系体积和样品浓度发生变化,影响了高通量的测定。本研究所建立的设备完善的高通量筛选平台可实现最高温度为70℃的恒温储藏箱对样品进行加热,因此我们尝试将茚三酮-铁与赖氨酸的反应温度下调到70℃,采用200mmol/L的赖氨酸溶液与茚三酮-铁试剂反应,每隔40min检测反应溶液在480nm处的吸光值,来观察测定结果的变化(数据未给出)。结果显示,在8h时抛物线坡度变缓,达到最高点,即200mmol/L的赖氨酸在70℃需要8h才能反应完全,虽然较好地降低了温度,准确度较高,但是由于时间较长,大大限制了高通量检测赖氨酸的可行性,所以此方法要想应用于高通量测定还需要进一步的改进。
4 结论
本研究对用于赖氨酸检测的茚三酮方法进行了改进,在pH2.2的磷酸氢二钠-柠檬酸缓冲液中只有赖氨酸可与茚三酮-铁试剂发生480nm处的特征显色反应,其最佳测定条件为100℃反应35min。改进的方法不仅可以排除杂氨基酸和有机酸的干扰,达到快速准确特异测定赖氨酸浓度的目的,而且在工业发酵中可作为简单可行的方法实现对赖氨酸的定性和定量分析。
[1] Zelder O, Klopprogge C, Schöner H, et al.L-lysine fermentation[J].Microbial Technolol, 2005, 1(1):211-240.
[2] Rao D, Razak SA, Praveena B, Swamy A.Dissolved oxygen concentration analysis of L-Lysine fermentation production by Corynebacterium glutamicum[J].The Internet Journal of Pharmacology,2008, 6(1), DOI:10.5580/19f0.
[3] Anastassiadis S.L-Lysine fermentation[J].Microbial Technolol,2007, 1(1):11-24.
[4] Nelofer R, Syed Q, Nadeem M.Statistical optimization of process variables for L-lysine production by Corynebacterium glutamicum[J].Türk Biyokimya Dergisi, 2008, 33(2):50-57.
[5] Debatosh D, Akshay B, Vidya C.Lysine:Is it worthmore?[J].Cytotechnology, 2001, 36:3-32.
[6] Oh JW, Kim SJ, Cho YJ, et al.Strain of corynebacterium glutamicum andmethod for producing L-lysine:US, 5268293[P].1993-12-7.
[7] Nakazawa M, Takahashi D, Onishi N, et al.L-Lysine fermentation:US, 20067012152[P].2006.
[8] Nakazawa M, Takahashi D, Onishi N, et al.Method for producing lysine derivative:US, 20067012152[P].2006.
[9] Emmert JL, Douglas MW, Boling SD, et al.Bioavailability of lysine from a liquid lysine source in chick[J].Poultry Science, 1999, 78(3): 383-386.
[10] 周伟.浅谈赖氨酸行业现状和发展趋势[J].发酵科技通讯,2007(6):31-35.
[11] 闫洪颖, 柳晓峰.2010年中国赖氨酸市场回顾及2011年展望[J].中国畜牧杂志, 2011, 47(4):23-30.
[12] 王镜岩, 朱圣庚, 徐长法.生物化学[M].北京:高等教育出版社, 2002:138-139.
[13] Friedman M.Applications of the ninhydrin reaction for analysis of amino acids, peptides, and proteins to agricultural and biomedical sciences[J].Agric Food Chem, 2004, 52:385-406.
[14] Dietzen DJ, Weindel AL, Carayannopoulos MO, et al.Rapid comprehensive amino acid analysis by liquid chromatography/tandemmass spectrometry:comparison to cation exchange with post-column ninhydrin detection[J].Rapid Commun Mass Spectrom, 2008, 22(22):3481-3488.
[15] Chinard FP.Photometric estimation of proline and ornithine[J].J Biol Chem, 1952, 91(5):91-95.
[16] Hsieh CL, Hsiung KP, Su JC.Determination of lysine with ninhydrin-ferric reagent[J].Analytical Biochemistry, 1995, 224(1):187-189.
[17] 杨菁, 孙黎光, 白秀珍, 周海涛.异硫氰酸苯酯柱前衍生化反相高效液相色谱法同时测定18种氨基酸[J].色谱, 2002,20(4):369-371.
[18] Beckwith AC, Paulis JW, Wall JS.Direct estimation of lysine in cornmeals by the ninhydrin color reaction[J].Food Chem, 1975,23(2):194-196.
[19] 刘飞飞, 李群, 于岚.茚三酮比色法定量检测赖氨酸条件的研究[J].中国食品添加剂, 2010, 5:223-234.
