Prader-Willi 综合征Ⅰ型和Ⅱ型缺失新生儿表型分析
2013-09-10樊子川程国强王来栓周文浩
梅 枚 杨 琳 樊子川 程国强 王来栓 曹 云 陆 炜 周文浩
Prader-Willi 综合征( PWS) 是一种累及多系统的非孟德尔遗传性疾病,是印记遗传的典型代表,人群发病率为1/10 000 ~1/30 000[1~3]。PWS 在新生儿期以严重肌张力低下和吮吸障碍为特点,随后可出现过度饮食、肥胖,并常伴有特殊面容、动作语言发育迟缓、性腺发育不良、认知障碍和行为异常等。PWS 发病机制包括:①父源15q11-13 区域缺失占65% ~75%;②15q11-13 母源单亲二倍体( UPD)占20% ~30%;③印迹基因功能缺陷占1% ~3%[4]。在父源性缺失病例中,远端断裂点常固定,而依据近端断裂点不同,可分为Ⅰ型缺失( del Ⅰ) 、Ⅱ型缺失( del Ⅱ) ,缺失大小分别约为6. 58 和5. 33 Mb[5]。1993 年,Holm 等[6]在Pediatrics 上发表了PWS 临床诊断标准,但其确诊依赖于遗传学检测,尤其是对于临床表现不典型的新生儿。在基因型和表型关联性研究方面,已有的研究数据显示: UPD 型,父源性Ⅰ型缺失、Ⅱ型缺失患者可有表型异常[7~12],但以往研究涉及新生儿的较少。詹实娜等[13]对9 例父源性缺失型、4 例UPD 型PWS 新生儿研究时发现,UPD 型患儿的特殊面容和男性生殖器发育不全的发生率低于父源性缺失型患儿,但对Ⅰ、Ⅱ 型缺失未做比较。本文纳入经Affymetrix SNP 芯片筛查全基因组拷贝数变异( CNVs) 或甲基化多重连接探针扩增技术( MS-MPLA) 确诊的PWS 父源性缺失患儿,分析Ⅰ、Ⅱ型缺失临床表型的差异,丰富PWS表型基因型的数据。
1 方法
1.1 对象 纳入2011 年9 月至2012 年12 月复旦大学附属儿科医院新生儿科符合PWS 临床诊断[6]的患儿,经Affymetrix SNP 芯片筛查全基因组拷贝数变异或MS-MLPA确诊和分型为父源性Ⅰ、Ⅱ型缺失患儿。
1.2 MLPA MLPA 检测试剂盒SALSA ME028 购自MRCHolland 公司,覆盖15q11-13 区域,并包含可检测不同甲基化水平的探针。操作依照产品说明书,DNA 预变性后与MLPA 探针杂交,左右探针经过链接反应后加入通用引物扩增。扩增产物经ABI 3500xl DNA 分析仪进行毛细管电泳。之后使用GeneMarker 软件对电泳产物片段长度和峰值进行分析,并通过与对照DNA 的比较得到拷贝数水平。
1.3 SNP 芯片检测 使用NanoDrop ND-1000 分光光度计检测DNA 浓度及吸光度值,电泳检测DNA 降解程度。取基因组DNA 3 ~5 μg 进行芯片检测。采用Affymetrix 公司Cytogenetic Whole-Genome 2.7 M 芯片进行检测。结果采用Chromosome Analysis Suite ( ChAS) 软件进行数据结果的分析。除外假阳性结果、在 DGV ( Database of Genomic Variation) 中报道的为正常人群的CNVs 和片段区域内不包含基因的CNVs。
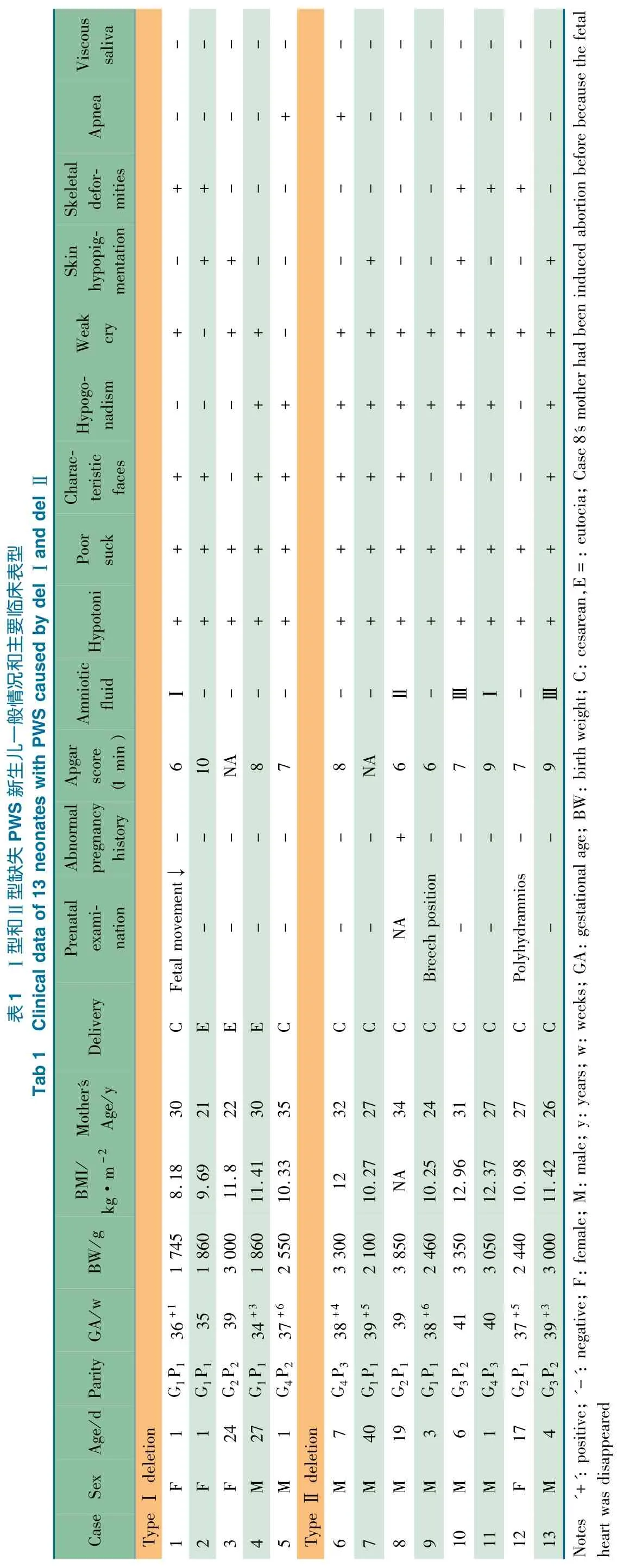
1.4 临床资料截取 ①一般情况: 性别、胎龄、出生体重、入院年龄、体重、身长、Apgar 评分、母亲年龄、健康状况、产前检查、羊水情况、分娩方式和异常家族史。②临床表型:中枢性肌张力和吸吮力,喂养困难程度,特殊面容,生殖系统发育情况,哭声,色素减低情况和嘴角结痂等;X 线胸片,超声心动图,头颅MRI/CT,血、尿串联质谱和甲状腺功能等实验室和影像学表现。
1.5 统计学方法 计量资料以x±s表示,计数资料以百分比表示,对Ⅰ、Ⅱ型缺失患儿的临床表型数据进行描述分析。
2 结果
2.1 一般情况 13 例父源性缺失PWS 新生儿进入分析,其中MS-MLPA 诊断10 例( 图1) ,3 例采用Affymetrix SNP芯片筛查诊断。如表1 所示,Ⅰ型缺失5 例,男2 例,女3例。平均入院年龄10.8 d; 平均胎龄36.2( 34 ~39) 周; 出生体重( 2 203 ±548) g,入院BMI 10.2 ±1.5。母孕年龄(27.6 ±5.9) 岁。1 min Apgar 评分( 7.8 ±1.7) 分,3 例早产,2 例剖宫产;例1 有产前胎动减少和羊水污染表现;5 例均无异常家族史。Ⅱ型缺失8 例,男7 例,女1 例。平均入院年龄12.1 d; 平均胎龄38.9( 37 ~41) 周; 出生体重(2 943 ±578) g,入院BMI 11.5 ±1.0。母孕年龄( 28.5 ±3.4) 岁。1 min Apgar 评分(8.8 ±1.2) 分;8 例均为足月剖宫产,4 例羊水污染,例8 母亲孕期存在无胎心妊娠史。
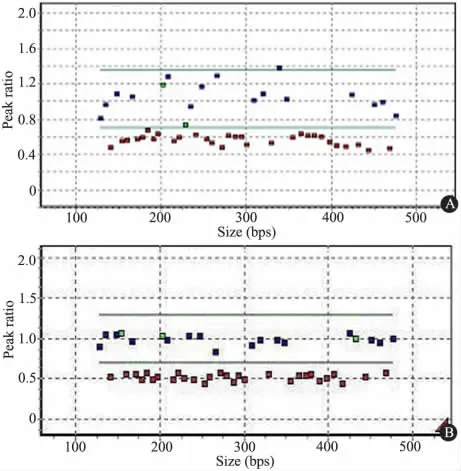
图1 Ⅰ型缺失和Ⅱ型缺失MS-MLPA 结果Fig 1 MS-MLPA's result of del Ⅰand del Ⅱ
2.2 两种缺失型临床表型 表1 显示,Ⅰ型和Ⅱ型缺失各表型的发生率:肌张力低下和吸吮力差均为100%,特殊面容分别为80%(4 例) 和50%(4 例) ,色素减退分别为40%(2 例) 和37.5%(3 例) ,生殖系统异常分别为40%(2 例)和87.5%(7 例) 。5 例伴有骨关节发育异常( Ⅰ型2 例、Ⅱ型3 例) 。Ⅰ和Ⅱ型缺失各有1 例发生呼吸暂停( 例5 和例6) 。Ⅱ型缺失中例12 伴腭裂。13 例均未发现有唾液黏稠导致嘴角结痂现象。
其他临床表型:12/13 例X 线胸片均未见明显异常。10/13 例行超声心动图检查,例1( Ⅰ型缺失) 提示为房间隔缺损,例9( Ⅱ型缺失) 提示为房间隔缺损、室间隔缺损。Ⅰ型缺失4/5 例行头颅CT 或MRI 检查,显示例1 和3 有颅内出血,例5 为左侧侧脑室增宽;Ⅱ型缺失7/8 例行头颅CT/MRI 检查,例9 提示双侧侧脑室饱满,胼胝体略薄,例13 部分脑外间隙增宽,例7 髓鞘化延迟,余4 例未发现异常。例1( Ⅰ型缺失) 行腹部B 超检查发现双肾多发囊性占位; 例2( Ⅰ型缺失) 尿串联质谱提示二羧酸尿可能; 例4( Ⅰ型缺失) 眼底检查示双眼色素淡,眼底呈“晚露”状改变,可见脉络膜血管;例9( Ⅱ型缺失) 喉镜检查示先天性喉软骨发育不良。
3 讨论
PWS 父源性缺失病例中,远端断裂点常固定( BP3) ,I 型缺失的近端断裂点在BP1,Ⅱ型缺失的近端断裂点在BP2。BP1-BP2 区段大小约500 kb,包含4 个保守基因NIPA1、NIPA2、CYFIP1 和GCP5,其中NIPA1 和脑发育密切相关[14]。以往关于基因型和表型关联性分析主要集中在父源性缺失型和UPD 型,涉及两种缺失型的较少。2004 年,Butler 等[8]对12 例Ⅰ型缺失和14 例Ⅱ型缺失青年患者进行研究后发现,两种缺失型患者的体格测量指标无明显差异,但Ⅰ型缺失患者的行为和智力问题较Ⅱ型缺失严重,可能和4 个保守基因有关。Milner等[15]也发现,Ⅰ型和Ⅱ型缺失患者临床表型无明显差异,但Ⅰ型缺失患者总体能力水平更低。Varela 等[16]发现,Ⅰ型缺失患者语言发育较Ⅱ型缺失更落后。上述研究对象为大年龄儿童和成人,目前国际上尚无Ⅰ、Ⅱ型缺失PWS 新生儿表型差异的数据。对于新生儿行为、语言及心理问题尚无有效判定方式,其他表型上是否具有差异尚无数据支持。
表1 数据显示,Ⅰ型缺失患儿的平均胎龄及出生体重较Ⅱ型缺失小。早产在Ⅰ型缺失组中的发生率为60%,在Ⅱ型缺失中的发生率为0,提示Ⅰ型缺失患儿可能更易发生早产。而胎动减少、羊水污染和胎位异常在分型判断上缺乏一定的指向性。
本文13 例缺失型PWS 新生儿主要临床表现方面,肌张力低下和吸吮力差发生率达100%,其后依次为哭声减弱( 84.6%) 、生殖器异常(69.2%) 、特殊面容( 61.5%) 、骨关节异常( 38. 4%) 、色素减退( 38. 4%) 、呼吸暂停(15.4%) 和腭裂(7.7%) 。肌张力低下和吮吸力差是两型的共同表现,发生率为100%,与Gunay-Aygun 等[17]研究结果类似。Ⅰ型缺失较Ⅱ型缺失可能更易表现为特殊面容( 80% vs 50%) ,Ⅱ型缺失较Ⅰ型缺失生殖系统异常发生率高( 40% vs 87.5%) ,色素减退发生在Ⅰ、Ⅱ型缺失间相近;提示特殊面容和生殖系统异常可能对于区分Ⅰ型和Ⅱ型缺失有一定的提示作用。本文病例同时发现,4 例影像学检查提示可能存在发育异常( 左侧侧脑室增宽,双侧侧脑室饱满、胼胝体略薄,部分脑外间隙增宽,髓鞘化延迟) ,其中Ⅰ型和Ⅱ型缺失分别为1 和3 例,这些影像学异常是否具有临床意义尚需要随访研究支持。此外,PWS 新生儿的表型还涉及循环系统、泌尿系统、呼吸系统和代谢异常,如表现为房间隔缺损和( 或) 室间隔缺损、双肾多发囊性占位、二羧酸尿、喉软骨发育不良等,其中Ⅰ型缺失可能比Ⅱ型缺失有更多样的表型。
综上,肌张力低下和吸吮力差是PWS 新生儿共同的临床特征,Ⅰ型缺失可能更易发生早产,特殊面容,Ⅱ型生殖系统异常发生率较高,Ⅰ型缺失可能具有多样的临床表型,对其中涉及的基因机制仍有必要进行深入研究。
[1]Holm VA, Cassidy SB, Butler MG, et al. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics,1993,91(2):398-402
[2]Whittington JE, Holland AJ, Webb T, et al. Population prevalence and estimated birth incidence and mortality rate for people with Prader-Willi syndrome in one UK Health Region. J Med Genet,2001,38(11):792-798
[3]Vogels A, Van Den Ende J, Keymolen K, et al. Minimum prevalence, birth incidence and cause of death for Prader-Willi syndrome in Flanders. Eur J Hum Genet,2004,12(3):238-240
[4]Cassidy SB, Schwartz S, Miller JL, et al. Prader-Willi syndrome. Genet Med,2012,14(1):10-26
[5]Butler MG, Fischer W, Kibiryeva N, et al. Array comparative genomic hybridization (aCGH) analysis in Prader-Willi syndrome. Am J Med Genet A,2008,146(7):854-860
[6]Holm VA, Cassidy SB, Butler MG, et al. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics,1993,91(2):398-402
[7]Butler MG, Sturich J, Myers SE, et al. Is gestation in Prader-Willi syndrome affected by the genetic subtype? J Assist Reprod Genet,2009,26(8):461-466
[8]Butler MG, Bittel DC, Kibiryeva N, et al. Behavioral differences among subjects with Prader-Willi syndrome and type I or type II deletion and maternal disomy. Pediatrics,2004,113(3 Pt 1):565-573
[9]Gillessen-Kaesbach G, Robinson W, Lohmann D, et al.Genotype-phenotype correlation in a series of 167 deletion and non-deletion patients with Prader-Willi syndrome. Hum Genet,1995,96(6):638-643
[10]Cassidy SB, Forsythe M, Heeger S, et al. Comparison of phenotype between patients with Prader-Willi syndrome due to deletion 15q and uniparental disomy 15. Am J Med Genet,1997,68(4):433-440
[11]Butler MG. Hypopigmentation: a common feature of Prader-Labhart-Willi syndrome. Am J Hum Genet,1989,45(1):140-146
[12]Honea RA, Holsen LM, Lepping RJ, et al. The neuroanatomy of genetic subtype differences in Prader-Willi syndrome. Am J Med Genet B Neuropsychiatr Genet,2012,159B(2):243-253
[13]Zhan SN(詹实娜),He XY,Wang CZ, et al. Clinical phenotype study of Prader-Willi syndrome in 13 neonates. Chin J Evid Based Pediatr(中国循证儿科杂志),2012,7(3):200-204
[14]Chai JH, Locke DP, Greally JM, et al. Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am J Hum Genet,2003,73(4):898-925
[15]Milner KM, Craig EE, Thompson RJ, et al. Prader-Willi syndrome: intellectual abilities and behavioural features by genetic subtype. J Child Psychol Psychiatry,2005,46(10):1089-1096
[16]Varela MC, Kok F, Setian N, et al. Impact of molecular mechanisms, including deletion size, on Prader-Willi syndrome phenotype: study of 75 patients. Clin Genet,2005,67(1):47-52
[17]Gunay-Aygun M, Schwartz S, Heeger S, et al. The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics,2001,108(5):E92
