非光气法合成二苯基甲烷二异氰酸酯的研究进展*
2013-09-01王庆印康武魁李子健杨先贵刘绍英王公应
王庆印,康武魁,李子健,杨先贵,刘绍英,王公应
(1.中国科学院成都有机化学研究所,四川 成都 610041;2.中国科学院大学,北京 100049;3.青岛农业大学 化学与药学院,山东 青岛 266109)
二苯基甲烷二异氰酸酯(MDI)是一种极其重要的有机异氰酸酯,主要用于制造聚氨酯弹性体、聚氨酯硬质、半硬质泡沫塑料等。MDI凭借其自身的优点,迅速渗入各类聚氨酯产品,是目前世界上产量最大、用途最广的异氰酸酯[1,2]。
目前MDI的生产均采用传统工艺光气法。该生产工艺虽然成熟,但存在光气易挥发、剧毒、环境污染大,副产物氯化氢对设备腐蚀严重,造成生产装置造价昂贵、技术要求复杂、产品中含氯化物不易分离等缺点[3]。随着人们对绿色化学认识的提高,MDI的光气法生产工艺终将被淘汰,因此非光气法生产MDI已经成为国内外学者的研究热点之一,并相继开发了多种非光气法生产工艺路线。
本文综述了国内外非光气法(如三光气法、酯交换法以及氨基甲酸酯热分解法)合成MDI的研究进展。
1 三光气法
三光气[化学名为双(三氯甲基)碳酸酯(C3Cl6O3),简称 BTC]是一种稳定的固体化合物,广泛用于医药、农药、染料、颜料及高分子材料的合成,几乎可以替代光气、氯甲酸三氯甲酯的所有反应[4]。
BTC法制备MDI的合成合成路线见Scheme 1[5]。夏敏等[6]利用 BTC 代替传统的光气进行羰基化反应合成MDI,收率87%。研究结果表明,pH值对MDI收率的影响较大,pH 11~12时收率最大,高于或低于这个范围,收率均会降低。
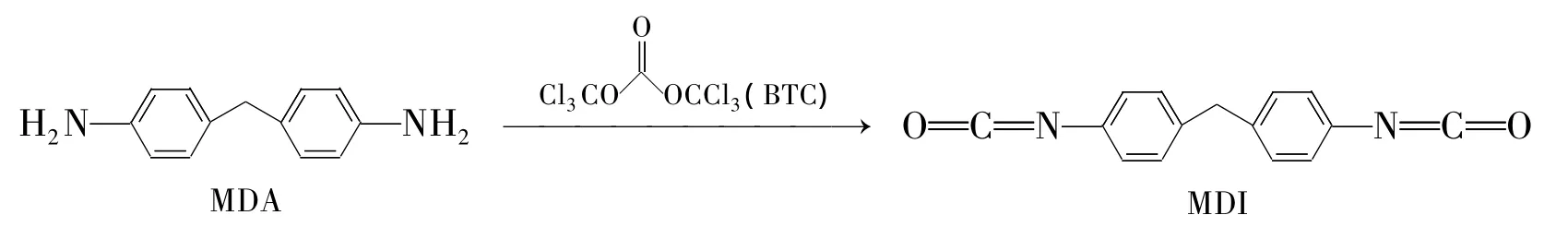
Scheme 1
利用BTC代替光气制备MDI,虽然具有使用方便、固体可称量、反应条件温和、收率高等优点,但是采用BTC法时,仍然会产生大量的盐酸,腐蚀设备,产品中含有氯化物的问题也没解决[7]。此法工业化前景暗淡,研究者对该法的研究也不多。
2 酯交换法
Frank等[8]开发了一种酯交换法合成MDI的工艺(Scheme 2)。首先苯氨基甲酸甲酯与甲醛在氯化锌的盐酸水溶液催化作用下,反应20 min,然后加入氯苯水洗两次,再加入NaHCO3水洗两次,过滤干燥得到4,4'-二苯甲烷二氨基甲酸甲酯(MDM)。将一定量的MDM溶于过量的苯异氰酸酯中,加入硅胶负载的SnCl4催化剂,升温至70℃,减压至0.4 kPa反应,精馏制得纯MDI。该反应的致命弱点是副产物多、成本高,因此工业化应用受到限制。
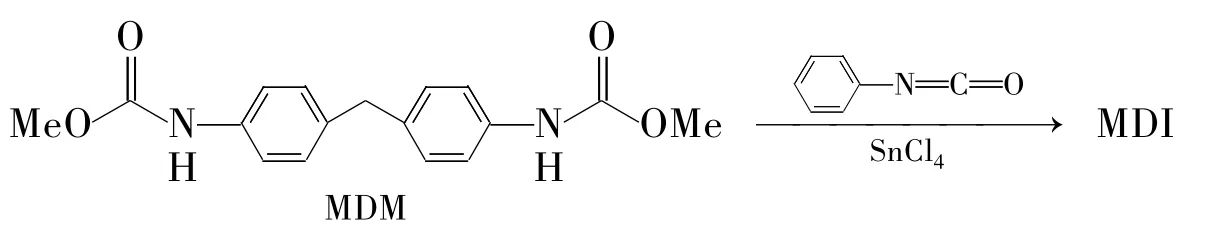
Scheme 2
3 氨基甲酸酯热分解法
由于初始原料的不同,使热分解合成MDI的方法呈多样性(Scheme 3)。但殊途同归,这些方法的最后一步都要经过热分解才能最终生成MDI,完成整个工艺流程。因此将这些方法统称为“4,4'-二苯甲烷二氨基甲酸酯(MDC)热分解法(MDC法)”。
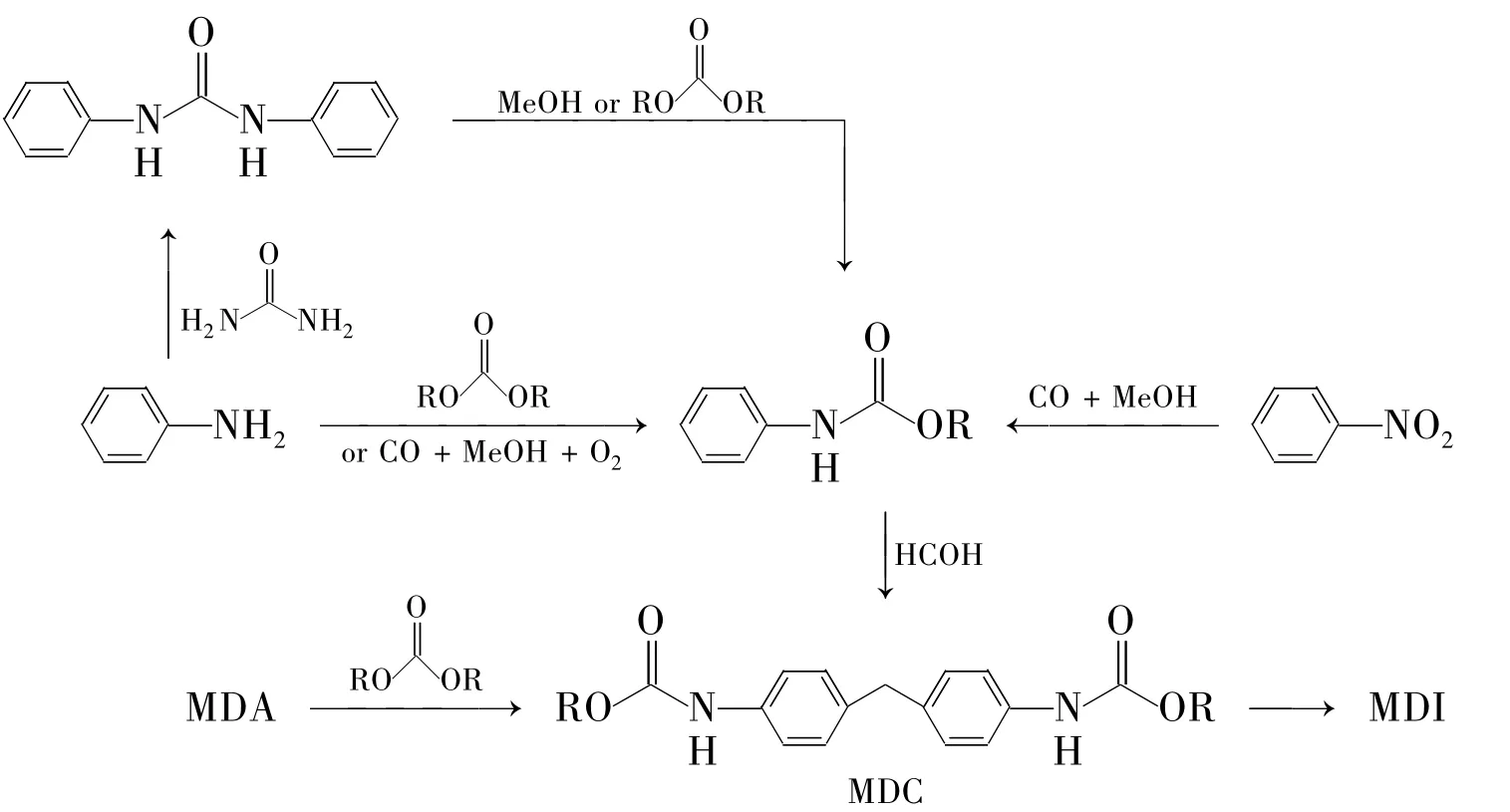
Scheme 3
MDC热分解是可逆吸热反应,为了在提高MDI收率的同时抑制副反应,关键环节是选择合适的反应器、优化工艺条件使产物及时从反应体系中分离,以及筛选优良的催化剂体系提高产物的选择性。下面主要从反应所采用的分解反应器和催化剂两方面介绍MDC热分解的研究。
3.1 热分解反应器
(1)管式分解反应器
汪多仁[9]采用两种不同的管式反应器进行热分解制备MDI。第一种采用石英管式反应器,将MDM溶于四氢噻吩溶液中,用泵以一定流速输入充满锌屑填料的石英反应器内,甲醇低温回收,粗产物于(90~95)℃/10 Pa蒸馏得到异氰酸酯混合物,MDI含量57%。第二种采用不锈钢降解器,在加热条件下,将4,4'-二苯甲烷二氨基甲酸乙酯(MDU,10%)的邻二氯苯溶液预热到(150~160)℃,从上向下以25 mL·min-1的速率喷入到不锈钢降解器内,降解器温度280℃,内充填拉西环,底部以3 NL·min-1速率通氮气,在1.5 MPa的压力下,连续降解制备异氰酸酯混合物,MDI含量 89.2%。
Jensen等[10]采用连续热解工艺,利用带隔板的管式反应器对低浓度的MDM热分解进行了研究,具体实验结果未见公开。Lewandowski等[11]采用Φ1.7 cm×90 cm的石英管作为热解反应器,管内填充铝条或固体催化剂,外部采用电热丝供热。将MDU(5%)的十二烷基苯溶液预热到120 ℃,以(0.3~1.5)g·dm-3·min-1的流量从反应管顶端导入,石英管加热到285℃,底部以(12~30)Ndm3·h-1的流量通入预热的氮气,反应时间100 min,MDU转化率98.1%,MDI选择性 87.2%。
Michael Arne[12]采用管式热分解塔,将 MDM(7%)的邻二氯苯溶液预热至250℃,用泵从顶端将反应液导入热分解塔中,让反应液从上向下流,同时从底部通入氮气,形成逆流接触,生成的副产物甲醇随氮气从底部排出,产物流入底部接收器中。在反应温度250℃,反应压力2 MPa,无催化剂作用下通过此反应器进行热分解,MDM转化率100%,MDI选择性93%。此工艺指标已经达到工业化的程度,可能主要是没有达到经济指标,因而未见工业化大生产。
Fukuoka等[13]采用 Φ2 cm ×400 cm 的管式反应器,填充物为铝制拉西环和狄克逊不锈钢填充物,将含有MDU(15%)的邻二氯苯溶液预热到160℃,以10 mL·min-1的流量从反应管顶端导入,反应管加热到270℃,底部以1.5 Ndm3·h-1的流量通入预热的氮气,反应压力0.8 MPa,反应液在低于100℃,减压条件下浓缩,含4,4'-MDI 87.5%,2,4'-MDI 10.7%和单边异氰酸酯1.8%。Shinohata等[14]也采用连续热解工艺,利用薄膜反应装置连续运转10 d,MDI收率99.5%。
李会泉等[15]采用流化床作为气相热解反应器,利用氮气作为载气将原料MDM输送进入流化床反应器,床料采用石英砂,反应温度450℃,压力为常压,反应物在反应器中的停留时间为1.5 s,MDI收率90%。采用流化床作为气相热解反应器,强化了反应过程的传热和传质,床层不宜结焦堵塞,但存在一定的返混。
利用管式反应器进行热分解的工艺,如果能避免使用大量的溶剂,节约能耗,该工艺会将非光气法生产MDI的研究推进到工业化阶段。
(2)釜式反应器
汪多仁[9]采用带搅拌器、温度计、压力计的五段充填式蒸馏塔,顶端有接收器及溶剂冷凝器、冷却管、接收器及预热装置、氮气吹入管、溶剂供给管的500 mL的耐压容器,向容器内投入邻二甲苯及一定量的DMU。通氮气将反应压力升至0.3 MPa,塔身预热到150℃,在反应过程中不断等量补加邻二甲苯,使系统内其总体积不变,于255℃反应90 min。于80℃出料,MDI收率95.1%。
王公应等[16]采用一锅法将MDM,溶剂和催化剂加入烧瓶中,在氮气氛围下热分解40 min,MDI收率63%。Takeuchi等[17]采用常压釜式反应器进行MDM热分解制得MDI,收率44.1%。Henson等[18]于 195 ℃反应 24 h 制得 MDI,收率46%。Miranda等[19]合成MDI的收率为97%。
张永富等[20]将釜式反应器与精馏装置相结合,边反应边提纯,有效避免了产品与原料的返混。刘波等[21]利用微波辅助进行反应,微波反应器功率为(60~250)W,微波加热保持温度为(150~280)℃,绝压为(0.7~2.7)kPa的条件下,反应后减压纯化,MDI收率>90%。
此工艺属于间歇操作,通过对底部容器加热,使反应液以沸腾状态在釜内回流,完成分解。此操作存在的问题:MDI是热敏性物质,生成的MDI不能及时从体系中分离,易发生返混,从而带来更多的副产物;间歇操作也不利于工业放大。
3.2 催化剂的应用
MDC在高温下即使不使用催化剂也能分解生成MDI,但是反应速率慢,副产物多。因此很有必要加入高效催化剂提高收率。目前文献中使用的催化剂主要分为金属单质及其氧化物和无机盐、有机化合物和硅类化合物。
(1)金属单质及其氧化物和无机盐
Rosenthal等[22]将 MDM 溶于惰性溶剂如十六烷中,于(230~285)℃热分解制备MDI,使用重金属及其氧化物等做催化剂提高了热分解反应速率,所用重金属主要是钼、钒、锰、铁、钴、铬、铜、镍。
陈东等[23]研发的超细 ZnO催化剂对 MDM热分解具有较好的催化活性,并发现ZnO粒径大小和颗粒团聚状况是影响催化活性的重要因素。当其粒径在(100~200)nm时,MDM的转化率99.1%,MDI总收率 79.8%。同时,该催化剂经五次使用后其活性没有发生明显的降低。说明该催化剂是一种性能优异的MDM分解反应催化剂,具有潜在的工业应用前景。在此研究的基础之上,如果能将原料浓度进一步提高,减少产品分离过程中的能耗,那么该工艺具有很好的开发价值。
Deng等[24]对此催化剂进一步研究,将离子液体作为热载体,在240℃下反应10 min,MDI收率84%。关雪等[25]又将ZnO与Zn复配制备了ZnO/Zn催化剂。ZnO/Zn复配催化剂的最佳质量比为1∶1,并对工艺条件进行了优化,结果表明在催化剂用量为溶剂质量的1.6%,原料用量为溶剂质量的2.5%,于250℃反应80 min,MDM的转化率99.2%,MDI收率67.3%。
张永富等[26]以ZnO/Zn混合物为催化剂,邻苯二甲酸二辛酯为溶剂,通过程序升温和程序减压操作,由MDM制得MDI,收率92.3%。他们还以 Al/Al2O3混合物为催化剂制得 MDI,收率91.9%[27]。由此可见,在 ZnO/Zn 催化剂作用下,MDI收率较高,催化剂的确有很高的催化活性;但由于在反应过程中使用了大量溶剂,MDI含量低于2%,要制纯品MDI需蒸掉大量溶剂,能耗高,对进一步产业化是个瓶颈,因此还需要做深入研究。
王公应等[16]开发了一种由MDM热分解制备MDI的超细氧化物催化剂。该催化剂为元素周期表中ⅠB,ⅡB,ⅢA,ⅣA的金属或非金属的超细氧化物及它们的复合超细氧化物。如采用ZnO/SiO2,于240℃反应40 min。MDM 转化率99.6%,MDI收率63.1%。
王建国等[28]将 ZnO担载在 SBA-15上制备了对MDM热分解具有较高催化活性的ZnO/SBA-15复合纳米催化剂,该催化剂颗粒分布均一,分散性好,芳香异氰酸酯转化率高,选择性好。在该催化剂的作用下,MDM的转化率100%,MDI收率84.3%。此专利描述的催化剂活性很高,但是对催化剂的稳定性及回收利用等问题没有阐述。如果该催化剂的稳定性好且回收利用率高,那么该催化剂在非光气法合成MDI的应用中将发挥巨大作用。
Koichi等[17]发现在热分解一步中催化效果较好的催化剂为:氯化锌、乙酸锌、氧化锌、环烷酸锌、硬脂酸锌和氯化铝。其中,以氯化锌催化剂为实例,采用二苯醚为溶剂,MDU 5%,真空压力(93.3~96.0)kPa,于253 ℃反应90 min,产物不断从反应体系中分离出来,溶剂在反应体系中循环,MDI收率 98%。Sydor等[29]曾利用 FeCl3做催化剂,研究MDM在400℃的反应器内气相热分解制备MDI。
王公应等[30]开发了一种由MDM热分解制备MDI的催化剂。该催化剂为元素铋的单质、氧化物、硫化物、卤化物或盐。向MDM(10%)的邻苯二甲酸二丁酯溶液中加入一定量的Bi2O3,在减压条件下,于260℃反应30 min,MDM转化率97.8%,MDI收率 82.8%。
金属单质及其氧化物和无机盐做热分解催化剂,反应体系一般为多相,催化剂易于回收利用。锌类催化剂特别是氧化锌的催化活性要高于其它金属氧化物。
(2)有机化合物
Henson等[31]在195℃条件下热分解 MDM(5%)制备MDI,其中N,N-二甲基苯胺既做溶剂又做催化剂,加快了MDM热分解的速率,反应24 h,MDI收率 46%。Shawl等[32]利用盐酸吡啶做催化剂,1,3,5-三甲基苯为溶剂,于161℃常压反应120 min由MDU合成MDI,MDU转化率99%,MDI选择性98%。Hammen等[33]采用二丁基二月桂酸锡为催化剂,将含MDU(50%)的环丁砜溶液以100 g·h-1的流量进入薄层蒸发器(Φ35 mm×300 mm,内置金属搅拌桨)进行反应,反应温度270℃,反应压力4 kPa。气相混合物经二级冷凝分为两部分:Ⅰ组份为溶剂和乙醇的混合物,Ⅱ组份为溶剂、MDI,MDU和MIU的混合物。Ⅱ组份经环己烷萃取四次后,单程收率54.1%,连续收率90%。Sundermann等[34]发明了一种在助剂作用下MDM热分解制备MDI的方法。反应温度(150~350)℃,反应压力(0.1~2 000)kPa,助剂为氯化氢、有机酸氯化物以及有机氯化锡(Ⅳ)。所用助剂不仅加快了反应速率,而并提高了MDI收率。
有机化合物做为热分解催化剂,反应体系一般为均相,催化剂与反应物混合均匀,但存在的问题是催化剂的稳定性差和回收利用难。
(3)硅类化合物
Uriz等[35]利用蒙脱石 K-10为催化剂,十氢化萘为溶剂,在190℃下反应24 h制得 MDI。MDM转化率接近100%,MDI收率68%。Miranda等[36]采用蒙脱石为催化剂,连续反应24 h制得MDI,收率97%。
硅类化合物做为热分解催化剂,其优点在于价格便宜,但是反应时间过长,不利于工业生产。
4 结束语
近年来,随着人们对于环保和安全的重视,非光气法生成MDI已经成为一个新的焦点。各种非光气法路线竞相开展,其中最有工业化前景的非光气法路线为氨基甲酸酯热分解路线。降低成本、实现连续化是此路线研究的难点和关键。
[1] 徐培林,张淑琴.聚氨酯材料手册[K].北京:化学工业出版社,2002.
[2] 余黎明,郑宝山.国内外二苯甲烷二异氰酸酯市场分析[J].化学工业,2008,26(7):46-51.
[3] 陈东,王公应,薛援,等.MDI清洁生产研究进展[J].天然气化工,2005,30(1):58-62.
[4] 徐子成,毛福华,张慧文,等.三氯甲基碳酸酯(三光气)的合成及其应用[J].上海化工,19(4):4-6.
[5] 高俊杰,李会泉,张懿,等.MDI清洁合成工艺研究进展[J].化工进展,2009,28(2):309-315.
[6] 夏敏,堂建新.二苯甲烷-4,4'-二异氰酸酯的非光气合成法[J].株洲工业学院学报,2000,14(3):1-2.
[7] 东玉武,孙建梅,翟江,等.三氯甲基碳酸酯替代光气的应用[J].天津化工,2002,6:41-42.
[8] Frank D Mango.Production of 4,4'-alkylidene diphenyl diisocyanate[P].US 4 163 019,1979.
[9] 汪多仁.非光气法生产 MDI[J].四川化工,1995,(3):57-59.
[10] Arne T Jensen.Process for the preparation of organic isocyanates[P].US 5 449 817,1995.
[11] Grzegorz Lewandowski,Eugeniusz Milchert.Thermal decomposition of methylene-4,4'-di(ethylphenylcarbamate)tomethylene-4,4'-di(pheny lisocyanate)[J].Journal of Hazardous Materials,2005,A119:19-24.
[12] Michael Arne.PEP Review 2004-2 MDI[J].http://www.ihscom/products/chemical/technology/pep/reviews/mdi.aspx
[13] Shinsuke Fukuoka,Masazumi Chono,Tomonari Watanabe,et al.Method of manufacture of diphenylmethane diisocyanates[P].US 4 547 322,1985.
[14] Masaaki Shinohata,Nobuhisa Miyake.Isocyanate production press[P].US 20 110 092 731Al,2011.
[15] 李会泉,李新涛,包炜军,等.一种气相热解制备异氰酸酯的系统及其方法[P].CN 201 010 174 969.2,2010.
[16] 王公应,陈东,冯秀丽,等.一种用于二苯甲烷二氨基甲酸酯分解制备二苯甲烷二异氰酸酯的超细氧化物催化剂[P].CN 200 510 021 147.X,2005.
[17] Koichi Takeuchi,Seiji Hasegawa,Shinobu Aoki.Process for preparing polymethylene polyphenyl polyisocyanate[P].US 4 307 029,1981.
[18] Thomas R Henson,John F Timberlake.Preparation of organic isocyanates[P].US 4 294 774,1981.
[19] Sergio Castillon Miranda,Carmen Claver Cabrero,Elena Fernandez Gutierrez,et,al.Isocyanate production procedure[P].US 6 639 101,2003.
[20] 张永富,管宪文,赵战如,等.一种氨基烷酸酯热解制备异氰酸酯的装置[P].CN 200 910 014 822.4,2009.
[21] 刘波,由军,王毅,等.微波辅助合成4,4'-二苯甲烷二异氰酸酯的方法[P].CN 201 010 129 064.3,2010.
[22] Rudolph Rosenthal,John G Zajacek.Catalytic production of isocyanates from esters of carbamic acids[P].US 3 919 279,1975.
[23] 陈东,刘良明,王越,等.氧化锌催化二苯甲烷二氨基甲酸甲酯分解反应[J].催化学报,2005,26(11):987-992.
[24] Youquan Deng,Xiaoguang Guo,Feng Shi,et al.Method for preparing isocyanates by liquid-phase thermal cracking[P].US 20 110 021 810,2011.
[25] 关雪,李会泉,刘海涛,等.ZnO/Zn复配催化剂热分解4,4'-二苯甲烷二氨基甲酸甲酯制备4,4'-二苯甲烷二异氰酸酯的研究[J].北京化工大学学报(自然科学版),2009,36(4):12-16.
[26] 张永富,王莉,管宪文,等.一种用于氨基烷酸酯热解制备异氰酸酯的复合催化剂[P].CN 200 910 014 820.5,2009.
[27] 张永富,冯柏成,管宪文,等.一种制备异氰酸酯的方法[P].CN 200 910 014 821.X,2009.
[28] 王建国,郭星翠,秦张峰,等.ZnO/SBA-15复合纳米催化剂及制法和应用[P].CN 101 269 342A,2008.
[29] Walter Joseph Sydor,Raritan N J.Process for converting urethanes to isocyanates[P].US 3 734 941,1973.
[30] 王公应,戴云生,王庆印,等.一种芳(烷)基氨基甲酸酯热分解制备异氰酸酯的催化剂及应用[P].CN 200 710 048 329.5,2007.
[31] Thomas R Henson,John F Timberlake.Preparation of organic isocyanates[P].US 4 294 774,1981.
[32] Edward T Shawl,Haven S Kesling.Process for the preparation of methylene diphenylene diisocyanates and polymethylene polyphenylenepoly(diisocyanates)[P].US 4 873 364,1989.
[33] Giinter Hammen,Hartmut Knofel,Wolfgang Friederichs.Process for the preparation of polyisocyanates[P].US 5 043 471,1991.
[34] Rudolf Sundermann,Klaus Konig,Theodor Engbert,et al.Process for the preparation of polyisocyanates[P].US 4 388 246A,1983.
[35] Pedro Uriz,Marc Serra,Pilar Salagre,et al.A new and efficient catalytic method for synthesizing isocyanates from carbamates[J].Tetrahedron Letters,2002,43:1673-1676.
[36] Sergio Castillon Miranda,Carmen Claver Cabrero,Elena Fernandez Gutierrez,et al.Isocyanate production procedure[P].US 6 639 101,2003.
