盐酸厄洛替尼的合成*
2013-09-01晏燕,王超,周莉,孙健
晏 燕,王 超,周 莉,孙 健
(1.中国科学院成都生物研究所天然产物研究中心,四川 成都 610041;2.中国科学院大学,北京 100049)
盐酸厄洛替尼[又名特罗凯,N-(3-乙炔基苯基)-6,7-二(2-甲氧基乙氧基)-4-喹唑啉胺盐酸盐(5)]是一种新型的治疗局部晚期转移性非小细胞肺癌的靶向药物[1~3],多项临床研究表明其疗效确切,毒副作用小,可以显著地减轻患者的痛苦,延长患者的寿命,并且在晚期胰腺癌、头颈部恶性肿瘤的治疗中也显现了一定的临床应用价值。其作用机制是在细胞内与底物竞争,抑制EGFR-TK磷酸化,阻断肿瘤细胞信号的转导,从而抑制肿瘤细胞生长,诱导其调亡[4]。
已报道的合成5的方法较多,但概括起来仅两种策略:一是通过4-取代喹唑啉(包括4-卤代、4-甲氧基、4-甲硫醚及4-酰氧基喹唑啉)与间胺基苯乙炔发生 SNAr反应制备[5~8];另一种是通过Dimroth重排合成[9]。两种策略均需使用价格较昂贵的间胺基苯乙炔为原料。
本文首次提出以6,7-二(2-甲氧基乙氧基)喹唑啉-4-胺(2)为关键中间体,以钯催化的Buchwald-Hartwig交叉偶联反应为关键步骤合成5。以4,5-二甲氧基乙氧基-2-胺基苯甲腈(1)为起始原料,经关环[10]、Buchwald-Hartwig 交叉偶联[11]、水解、BestmannOhira反应[12]和最后成盐合成 5(Scheme 1),总收率 34%。其结构经1H NMR和ESI-HR-MS确认。
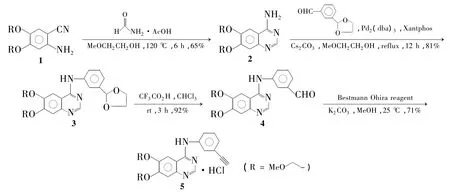
Scheme 1
该方法首次以2与乙二醇保护的间碘苯甲醛的Buchwald-Hartwig偶联构建C-N键,为5的合成提供了一条新颖的且具有工业化应用前景的途径。
1 实验部分
1.1 仪器与试剂
Avance Brucker 300 MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标);BioTOF Q型质谱仪。
HSGF254型薄层板,硅胶200目~300目,青岛海洋化工厂;其余所有试剂均为分析纯。
1.2 合成
(1)2的合成
氮气保护,在圆底三颈烧瓶中依次加入1 6.65 g(25 mmol),醋酸甲脒 7.81 g(75 mmol)和乙二醇甲醚50 mL,搅拌下于120℃反应6 h(TLC监测)。冷却至室温,过滤,滤液减压浓缩后用混合溶剂 A[V(二氯甲烷)∶V(甲醇)=20 ∶1]重结晶得灰白色固体 2 4.76 g,收率 65%;1H NMR δ:8.32(s,1H),7.63(brs,2H),7.62(s,1H),7.12(s,1H),4.18~4.15(m,4H),3.70~3.67(m,4H),3.28(s,6H);13C NMR δ:172.75,160.78,153.73,147.64,146.07,108.15,107.45,104.20,70.30,68.34,68.12,58.46;ESI-HR-MS m/z:Calcd for C14H19N3O4{[M+H]+}294.137 6,found 294.144 8。
(2)N-[3-(1,1-乙撑二氧甲基)苯基]-6,7-二(2-甲氧基乙氧基)-4-氨基喹唑啉(3)的合成
在圆底三颈烧瓶中依次加入2 4.40 g(15 mmol),乙二醇保护的间碘苯甲醛6.20 g(23 mmol),三(二亚苄基丙酮)二钯[Pd2(dba)3]0.82 g(0.9 mmol),4,5-双二苯基膦-9,9-二甲基氧杂蒽(Xantphos)1.04 g(1.8 mmol),碳酸铯 9.77 g(30 mmol)及乙二醇二甲醚50 mL,搅拌下氮气置换空气10 min;回流反应12 h(TLC监测)。缓慢冷却至室温,过滤,滤液浓缩后用混合溶剂A重结晶得黄色固体 3 5.76 g,收率 81%;1H NMR δ:9.55(s,1H),8.45(s,1H),7.98(d,J=8.1 Hz,1H),7.90(s,1H),7.79(s,1H),7.40(dd,J=8.0 Hz,7.8 Hz,1H),7.22(s,1H),7.17(d,J=7.6 Hz,1H),5.75(s,1H),4.30~4.27(m,4H),4.08~4.05(m,4H),3.78~3.37(m,4H),3.35(s,6H);13C NMR δ:156.34,153.59,152.91,148.09,146.88,139.50,138.46,128.34,123.03,121.66,120.07,108.93,108.14,103.19,102,82,70.15,70.09,68.38,68.06,64.84,58.44;ESIHR-MS m/z:Calcd for C23H27N3O6{[M+H]+}442.190 0,found 442.187 2。
(3)N-(3-甲酰基苯基)-6,7-二(2-甲氧基乙氧基)-4-氨基喹唑啉(4)的合成
氮气保护,在圆底三颈烧瓶中加入3 4.41 g(10 mmol)的三氯甲烷(50 mL)溶液,搅拌下于0℃慢慢滴加三氟乙酸1.48 mL(20 mmol),滴毕,于30℃反应3 h(TLC监测)。旋干溶剂得橙色固体 4 3.77 g,收率 92%;1H NMR δ:10.05(s,1H),8.68(s,1H),8.24(s,1H),8.13(d,J=8.0 Hz,1H),7.66(dd,J=8.0 Hz,7.8 Hz,1H),7.59(d,J=7.8 Hz,1H),7.41(s,1H),7.23(s,1H),4.33~4.27(m,4H),3.87~3.84(m,4H),3.47(s,3H),3.36(s,3H);ESI-HRMS m/z:Calcd for C21H23N3O5{[M+H]+}398.163 8,found 398.171 0。
(4)5的合成
氮气保护,在圆底三颈烧瓶中加入无水碳酸钾0.48 g(3.5 mmol)活化除水,搅拌下依次加入无水甲醇 25 mL,4 0.39 g(1 mmol),Bestmann Ohira试剂0.5 g(2.6 mmol),于25 ℃~30 ℃反应2 h。用硅藻土过滤,滤液旋干后用二氯甲烷(25 mL)溶解,依次用5%碳酸氢钠溶液(20 mL),饱和食盐水洗涤,分液,有机相用无水硫酸镁干燥,蒸除溶剂得厄洛替尼0.36 g,收率78%。用无水甲醇溶解,通入新制备的氯化氢干燥气体,于室温反应1.5 h。析出固体,过滤,滤饼用二氯甲烷洗涤,干燥得白色固体5 0.39 g,收率91%;1H NMR δ:11.42(s,1H),8.86(s,1H),8.38(s,1H),7.88(s,1H),7.78(d,J=8.0 Hz,1H),7.38~ 7.55(m,3H),4.32~ 4.38(m,4H),4.29(s,1H),3.77(m,4H),3.36(s,6H);ESI-HR-MS m/z:Calcd for C22H24N3O4Cl{[M+H]+}430.145 5,found 430.171 0。
2 结果与讨论
2.1 合成
(1)2的合成
2是该路线的关键中间体,文献报道的结构类似物的合成方法主要有4-卤代喹唑啉与氨水发生SNAr反应[13]和在微波辅助下,邻胺基苯甲腈与甲酰胺缩合关环[14]。实验中我们首先尝试了4-氯-6,7-二(2-甲氧基乙氧基)喹唑啉与浓氨水在甲醇中于60℃进行SNAr反应,但没能成功,可能是4-氯喹唑啉的活性比较低;选用微波辅助的方法,但是由于反应温度太高,原料和产品有部分分解,杂质多,收率低;最后借鉴邻胺基苯甲腈与醋酸甲脒缩合反应制备喹唑啉-4-胺的方法,发现1与醋酸甲脒于80℃~120℃反应就能高收率地得到2。
(2)3的合成
对于2与乙二醇保护的间碘苯甲醛的反应,我们分别考察了钯催化剂[Pd(OAc)2,PdCl2,Pd2(dba)3]和配体[2,2'-双二苯膦基-1,1'-联萘(BINAP),Xantphos和 1,1'-双(二苯基膦)二茂铁(dppf)]对偶联反应的影响,结果见表1。由表1可见,只有Xantphos有利于该底物的偶联反应,BINAP和dppf都没有效果。对于催化剂而言,Pd(OAc)2,PdCl2和Pd2(dba)3都能催化该偶联反应,Pd2(dba)3的效果最好,收率81%。

表1 催化剂和配体对Buchwald-Hartwig偶联反应的影响*Table 1 Effects of catalysts and ligands on Buchwald-Hartwig coupling reaction
[1] 王鹏,刘长庭,孙宝君,等.厄洛替尼治疗吉非替尼耐药的进展期非小细胞肺癌的临床观察[J].临床肿瘤学杂志,2010,15:617-620.
[2] 罗绍友,陈红,房芳,等.厄洛替尼治疗吉非替尼耐药老年非小细胞肺癌脑转移的疗效[J].临床荟萃,2012,27:301-303.
[3] 刘雨桃,郭继红,王燕,等.厄洛替尼治疗晚期非小细胞肺癌分类及回归树分析[J].中国肺癌杂志,2011,14:785-789.
[4] 李铭东,曹萌,吉民.盐酸埃洛替尼的合成[J].中国医药工业杂志,2007,38:257-259.
[5] 李付刚,陈毕峰.喹唑啉类抗癌药物的合成研究进展[J].精细与专用化学品,2010,18:45-53.
[6] Schnur R,Caughren.Alkynyl and azido-substituted 4-anilinoquinazolines[P].US 5 747 498,1998.
[7] Richard Bruce Brandon,Mervyn Rees Thomas.Polynucleotide marker genes and their expression for diagnosis of endotoxemia[P].WO 2 007 006 091,2007.
[8] 沈鑫,廖立新,林复兴.一种盐酸厄洛替尼的制备方法[P].CN 200 710 172 779,2009.
[9] Chandregowda V,Venkateswara Rao G,Chandrasekara Reddy G,et al.Convergent approach for commercial synthesis of gefitinib and erlotinib[J].Org Process Res Dev,2007,11:813-816.
[10] Sleebs B E,Czabotar P E,Fairbrother W J,et al.Quinazoline sulfonamides as sual binders of the proteins B-cell lymphoma 2 and B-cell lymphoma extra long with potent proapoptotic cell-based activity[J].J Med Chem,2011,54:1914-1926.
[11] Rauws T R M,Biancalani C,De Schutter J W,et al.Synthesis of new tetracyclic azaheteroaromatic cores via auto-tandem Pd-catalyzed and one-pot Pd-and Cu-catalyzed double C-N bond formation[J].Tetrahedron,2010,66:6958-6964.
[12] Quesada E,Raw S A,Reid M,et al.One-pot conversion of activated alcohols into 1,1-dibromoalkenes and terminal alkynes using tandem oxidation processes with manganese dioxide[J].Tetrahedron,2006,62:6673-6680.
[13] Marvania B,Lee P C,Chaniyara R,et al.Synthesis and antitumor evaluation of phenyl N-mustard-quinazoline conjugates[J].Bioorganic & Amp;Medicinal Chemistry,2011,19:1987-1998.
[14] Loidreau Y,Besson T.Microwave-assisted thermal decomposition of formamide:A tool for coupling a pyrimidine ring with an aromatic partner[J].Tetrahedron,2011,67:4852-4857.
