近红外发光Ag2S-CdS 核壳结构水溶性量子点的合成及光学性质
2013-08-20马云飞
成 戡 方 正 马云飞 张 华
(华东理工大学化学与分子工程学院,上海 200237)
直径小于10 nm 的半导体纳米晶通常被称为量子点(quantum dots),它们具有优异的,且依赖于尺寸、结构和组分的光学和电学性质,在太阳能电池、发光二极管(LED)、生物荧光标识以及生物传感等领域有潜在的应用价值[1-4]。同时,应用于生物领域中的量子点必须是生物相容的,因此,水溶性量子点的合成及性质也成为人们研究的热点之一[5-8]。在量子点成核或生长过程中掺入另一种过渡金属离子,根据掺入量和合成方法等差异性,可能得到掺杂量子点,合金或核壳结构等杂化量子点,这是一种可显著改变量子点光学和电学性质的有效方法,该领域工作也已引起了研究者们的广泛兴趣[9-11]。很多课题组通过各种方法合成了高质量的杂化量子点,实现了量子点在整个可见光范围的发光,为掺杂及核壳量子点的成核、生长和机理奠定了重要的理论基础[12-14]。CdS 毒性低,具有较大的Stocks 位移,可有效避免荧光自淬灭,更适于生物应用。Dong 等合成了柠檬酸盐修饰的水溶性CdS 量子点,对其掺杂Ag+后,荧光强度显著增强,在此原理上构筑了正比于Ag+浓度的离子传感器[15]。Yousefi 等用巯基乙醇作表面活性剂合成出CdS 及Ag 掺杂的水溶性量子点,结果同样表明掺入过渡金属可调控发光和吸收性质,获得了发射绿光的CdS 量子点[16]。Shen 等用微波辐射法合成了Ag 掺杂的CdS 量子点,量子产率高达58%,PL 在480~630 间[17]。以上报道的Ag 掺杂CdS 水溶性量子点光发射在650 nm 以内,而在近红外区域发光的量子点仍未见报道。近红外光在生物组织中有较深的穿透能力,可以实现光声成像,作为生物标识等[18]。单纯Ag2S(~1.0 eV,即1 240 nm)几乎不发光,但它与CdS (或掺杂CdS) 理论上可形成type-I 型核壳结构[19],当量子点吸收光后,CdS 导带中的光生电子和空穴将迁移到Ag2S 核中,减小了光生电子与空穴的能级差,可望表现出近红外发光特征。少量的Ag2S-CdS 复合纳米结构,如纳米线与纳米棒等已被报道[20-22],但实现Ag2S-CdS 核壳结构量子点的近红外发光仍是一个较大的挑战。
本工作以谷胱甘肽(GSH)作为表面活性剂,通过两步法,先生成Ag2S 核,再生长CdS,合成出高质量Ag2S-CdS 核壳结构水溶性量子点,实现了近红外发光,同时考察了不同实验条件,如Ag 含量,S含量及表面活性剂用量对量子点发光位置及强度的调控作用及机理。
1 实验部分
1.1 试 剂
醋 酸 隔(Cd(OAc)2,>98.5%),醋 酸 银(Ag(OAc),99.99%)购自Aldrich,还原谷胱甘肽(GSH,>98.0%),硫化钠(Na2S,>98.0%)购自上海化学试剂公司,所有药品并未作进一步纯化处理,实验过程中所用水均为去离子水。
1.2 实验方法
首先,将0.018 mmol Ag(OAc)和0.072 mmol GSH混合置于40 mL 去离子水中,在搅拌条件下用1.0 mol·L-1NaOH 将溶液pH 值调节到9.5,溶液置于100 mL 三颈烧瓶中,保持磁力搅拌并在N2保护下升温到85 ℃,随后向体系中加入含0.012 mmol Na2S 水溶液5 mL,反应4 min 后依次注入含0.012 mmol Na2S 水 溶 液5 mL 和 含0.02 mmol Cd(OAc)2水溶液20 mL,计时开始。间隔一定时间取样并监测样品的紫外吸收和荧光发射波长。反应结束后,将样品冷却到室温,并加入适量丙酮对量子点进行纯化,得到的固体沉淀物在氮气氛围中干燥,以得到纯净的固体。
1.3 产物分析和表征
将产物的水分散溶液多次滴在玻璃片上,干燥后在X 射线衍射仪上测定晶体结构 (型号:D8 Advance 德国Bruker 公司),该仪器配备的是固体二维探测器和新型的NaI 晶体闪烁计数器,辐射源为Cu Kα (λ=0.154 06 nm),加速电压及电流为40 kV和40 mA,样品测量范围为15°~90°。将样品的水溶液超声分散后滴在碳支持膜上,自然干燥后放入透射电子显微镜JEM-2010(日本电子)中测试所得样品的形貌(TEM),高分辨(HRTEM)及组分(EDS),加速电压为200 kV。紫外-可见吸收光谱(UV-Vis)和荧光发射光谱(PL)分别通过岛津UV-2450 紫外可见分光光度计和Varian 公司Cary Eclipse 荧光分光光度计获得。样品的室温荧光量子产率按参考文献的方法而获得[23],使用已知量子产率的荧光染料2-氨基吡啶(2-氨基吡啶溶于0.1 mol·L-1H2SO4,QY=60%)作为参照物。荧光寿命测试采用Edinburgh FLS 920光度计,激发光波长为370 nm,检测的荧光寿命范围为0~2 ms,并通过Edinburgh 公司提供的软件进行数据分析和处理。
2 结果与讨论
2.1 量子点结构及形貌
图1 所示为不同反应时间下所得样品的XRD图。由图中可看出,加入Cd 源前,产物结晶性较差,仅在约34°附近有个宽衍射包,对应于Ag2S(PDF No.11-0688)的(022)晶面,当加入Cd 源反应10min后,在26.3°,31.5°,43.7°,51.5°处各出现一个衍射峰,分 别 对 应 于 立 方 相CdS (PDF No.80-0019)的(111),(200),(220),(311)晶面,但对应于Ag2S 的衍射峰没有显著变化,随着反应时间延长,CdS 衍射峰位置无明显变化,但Ag2S 衍射峰逐渐分化,表明结晶性有所提高,直到2.5 h 后更加锐化。
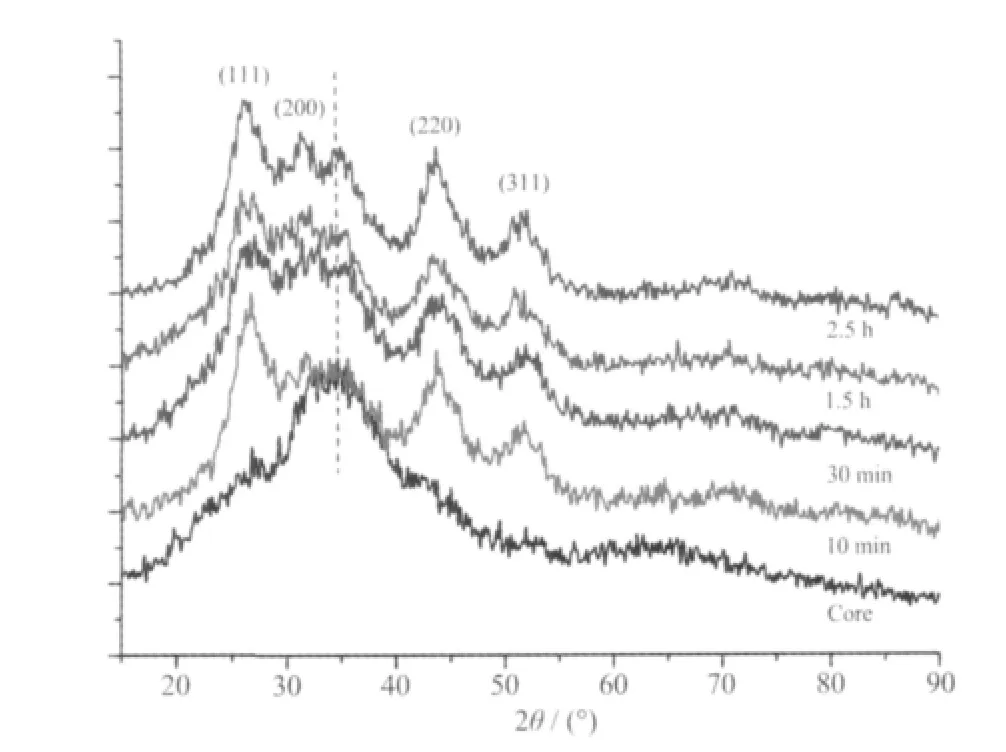
图1 不同反应时间所得样品的XRD 图Fig.1 XRD patterns of the samples prepared at different reaction times
图2 所示为核与不同反应时间所得量子点的TEM 和ED 图。在图2a 中看不到明显的衍射环,表明核晶化不好,与XRD 结果相符,核的直径约为3.2~5.1 nm。EDS 结果中Ag 与S 原子比为2.28,Ag过量,可能产物中有少量的Ag8S 存在,其(022)晶面衍射位于XRD 图中约34°位置。加入Cd 反应10 min 后,颗粒尺寸没有明显变化,但衍射强度明显增强,也与XRD 结果相符,同时Cd 含量增加(nAg/nCd=1.53),表明CdS 壳层开始生成,但Ag2S 物相仍占主导。值得一提的是,此时阳阴离子的原子比(nAg+nCd)/nS仅为0.82,说明已生成的核中有部分可能因为结晶性差和尺寸小等原因而溶解。反应时间为30 min时,量子点尺寸无明显变化,但单分散性更好,且Cd 及总的阳离子含量有所增加,nAg/nCd=0.74,(nAg+nCd)/nS=1.29,表明Cd 不断进入到量子点晶格中。1.5 h 时量子点的尺寸分布范围更小,为3.5~4.3 nm,Ag的相对含量有所增加,2.5 h 时量子点尺寸进一步地减小,为2.0~4 nm,晶粒边缘稍显不清晰。图2f 为30 min 样品的HRTEM 图,非常清晰的晶格条纹对应于CdS 的(111)晶面,但无法分辨核壳结构,其可能原因为Ag2S 核晶化程度低,且尺寸很小,这是核壳结构量子点所常见的特点。但根据我们的实验方案,体系在加入Cd 前先生成Ag2S 核,然后生长CdS,而Ag2S 的溶度积常数远大于CdS 的(Ksp(Ag2S)=6.3×10-50,Ksp(CdS)=8.0×10-27),反应体系中Ag 前驱物含量高,为Cd 的90%,即使有部分Ag2S 核被溶解或发生离子交换,最终产物中仍有大量Ag2S 核稳定存在,XRD 中也表明了此点。由此我们可推断出: 我们所得到的量子点是Ag2S-CdS 核壳结构,Ag大多分布于核中,下面的光学性质研究结果也说明了核壳结构的存在。
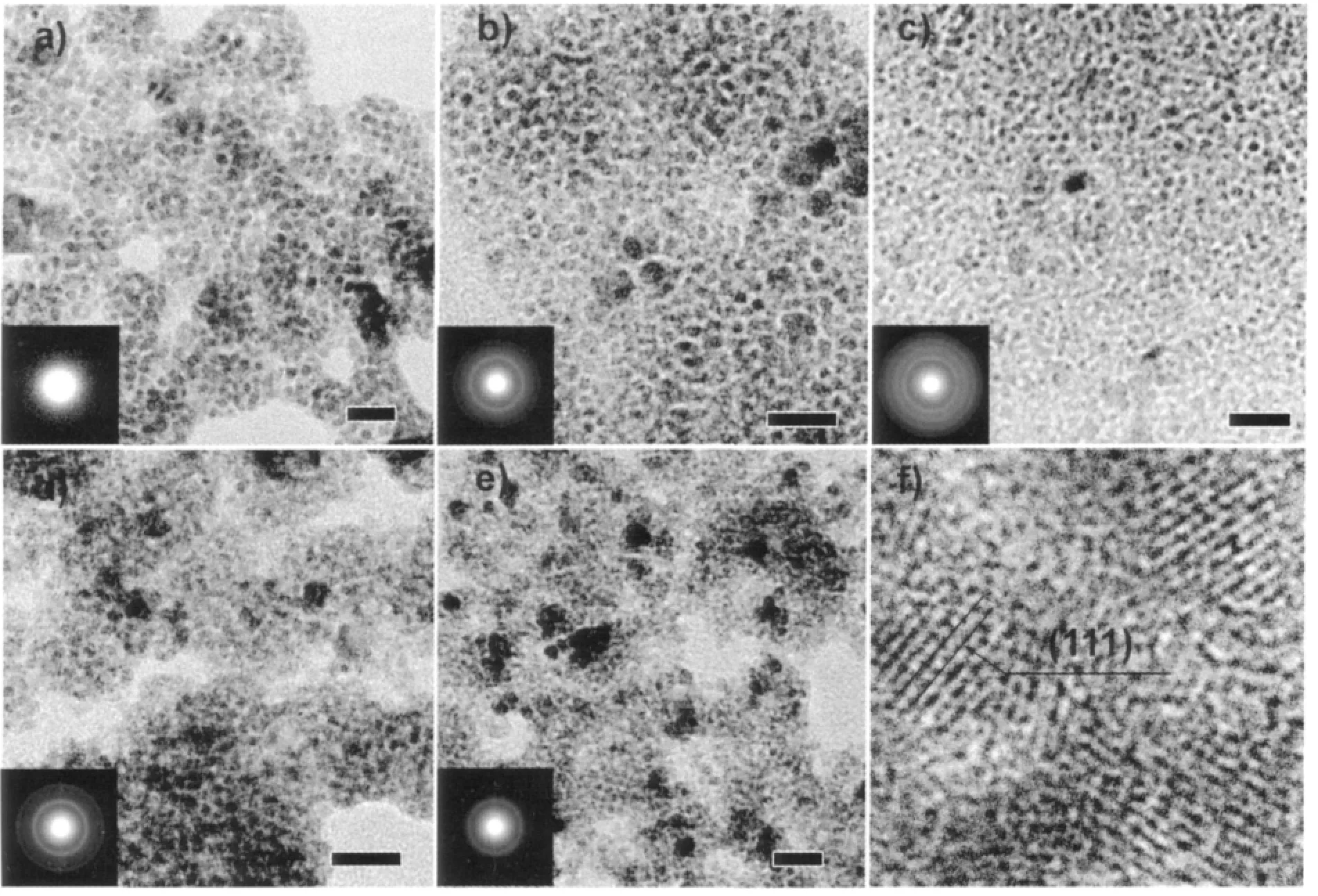
图2 Ag2S(a)核及不同反应时间所得量子点的TEM 和ED 图: (b) 10 min (c) 30 min (d) 1.5 h (e) 2.5 h,(f) 对应于c 中样品的HRTEM 图Fig.2 TEM and ED images of Ag2S core (a) and QDs prepared at different reaction times: (b) 10 min (c) 30 min(d) 1.5 h (e) 2.5 h, (f) HRTEM image of the sample shown in (c)
2.2 量子点的光学性质
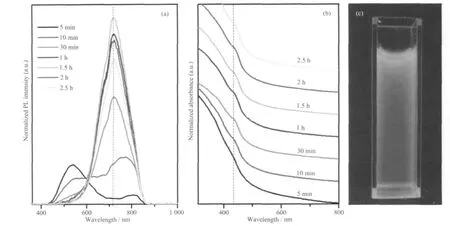
图3 Ag2S-CdS 核壳结构的PL (a), UV (b)及数码相机拍摄的紫外灯下的发光图(c)Fig.3 PL emission (a), UV-Vis absorption (b) spectra of the as-prepared Ag2S-CdS core-shell QDs and the digital image (c)
我们对所合成的量子点进行了UV-Vis 和PL的光学性质研究,结果如图3 所示。当注入Cd 源反应5 min 后,出现一个较强的位于540 nm(定义为峰1)和一个较弱的位于806 nm(定义为峰2)的宽发光峰,继续反应5 min 后,峰1 红移到552 nm,且强度有所下降,而峰2 蓝移到764 nm,强度显著增加,与短波长的峰强度比值由原来的0.23∶1 变为1.79∶1,随着反应时间进一步延长到30 min,则两个峰的位置发生很大变化,分别位于687 和714 nm,后者峰更强,而前者为一肩峰,较弱,两峰不断增强直到1.5 h 达最大值,714 nm 峰有较大的荧光寿命611 ns,687 nm 峰荧光寿命为532 ns,随后强度逐渐下降。值得注意的是,本工作中通过生长Ag2S-CdS 核壳结构,可使量子点在近红外区域发光,且在强度达最大值时,发光峰的半峰宽很窄,为150 nm,与文献报道中Ag 掺杂CdS 量子点的数值相似[9,17],表明量子点的尺寸分布小,电镜结果也已直观说明,且量子产率较高,为10%,从图3c 中非常明亮的红色发光也可知我们得到了高质量的近红外发光的量子点。另外,从UV-Vis 中(图3b)可以看出,反应时间为5 min 时没有明显的吸收峰,反应时间为10 min时,440 nm 处开始出现吸收峰,随着反应时间进一步延长,吸收峰位置没有显著变化,但峰形更加明显,可能与结晶性和单分散性提高相关。由Ag(量较Cd 少),Cd,S 形成的三元复合半导体量子点中,Ag元素的分布大致有3 种情况:第一,密集于中心,紫外吸收显示一个峰,外层大多为CdS,量子点稳定;第二,密集于壳的外层而具有较差的稳定性;第三,浓度由中心向外,先增大再减小,两个吸收峰是其显著特点[17]。在我们的实验中,首先生成Ag2S 核,当溶液中加入Cd 源后,在核的表面生长CdS 壳层,同时,Ag 有优异的扩散性(即使在室温下)[24],且结晶性差导致核活性高,不易稳定,因此,部分Ag 离子从核中通过界面扩散到壳层中[21,25],Cd 离子向核中扩散,实现阳离子交换。另一方面,Ag2S 的溶度积常数远大于CdS 的,Ag 前驱物含量高,即使有部分Ag2S核被溶解或发生离子交换,最终产物中仍有大量Ag2S 核稳定存在。因此,从以上的XRD,TEM,ED 及光学性质结果中,我们可推断出:(1) 我们所得到的量子点是Ag2S-CdS 核壳结构,Ag 大多分布于核中;(2) 在反应初始阶段,Ag 离子没有足够时间扩散,壳层中主要是较纯的CdS,PL 峰位于540 nm,来源于表面态或表面缺陷,这与本课题组以前的实验结果相符[26],而位于800 nm 附近的发光可能来自于更复杂的Cd 或S 空位[27];(3) 随着反应的进行,Ag 离子逐渐扩散到壳层,形成了Ag 掺杂的CdS 壳层,540 nm 处发光消失,缺陷相关的近红外发光占主导地位,并不断增强;(4) 当阳离子扩散与交换达平衡时,发光强度最大,反应时间进一步延长时,壳层中Ag 含量随之增加并向表层迁移和输运,到一定程度后产生过多的表面缺陷而使发光强度降低。根据type-Ⅰ型核壳结构发光机理[28-29],Ag2S-CdS 核壳结构量子点可实现较强的近红外发光的另一个可能原因是: 当量子点吸收光后,CdS 导带中的光生电子跃迁到Ag2S 核中,减小了光生电子与空穴的能级差,进而表现出近红外发光特征。相关近红外发光机理仍值得进一步研究。但无论如何,本工作已成功获得了高质量的Ag2S-CdS 核壳结构量子点,实现了很强的近红外区发光,且稳定性较好,所得量子点用紫外灯连续光照60 h 后,荧光强度没有明显减弱,可望应用于生物成像与标识中。
2.3 不同含量Ag 前驱物对光学性质的影响
在复合半导体量子点中生成掺杂,合金或核壳结构与两种金属源物质的比例密切相关。图4 为加入不同含量Ag 前驱物时所得样品的PL 谱图及强度对比图。当Ag 含量仅为5%时(nCd∶nAg=20∶1),只在~588 nm 处有较明显的发光峰,当Ag 含量增大到15%时(nCd∶nAg=20∶3),量子点在500~750 nm 间有很宽的发光平台,表现出了白光特征,同时714 nm处出现一明显发光峰,当Ag 含量继续增大到30%时(nCd∶nAg=20∶6),714 nm 峰稍有红移至722 nm,并显著增强,682 nm 处肩峰出现,600 nm 以内的峰强度逐渐下降,随着Ag 含量进一步增加,肩峰的近红外发光谱强度继续增强,直到Ag 含量为90%时(nCd∶nAg=20∶18)达最大值。当Ag 含量大于Cd 时,量子点中由Ag 引入的缺陷越来越多,导致荧光强度有所下降。图4b 中为近红外发光强度随Ag 含量和反应时间的变化关系。由此可知,改变Ag 含量可调控量子点的发光位置和强度,当很低时,其主要分布在核中,壳层主要由CdS 组成,因此表现出CdS 的发光特性,随着Ag 含量不断增加,壳层中Ag 含量也不断增加,进而实现近红外发光。
2.4 不同S 源的量对光学性质的影响
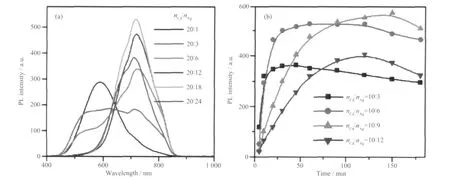
图4 加入不同含量Ag 前驱物时所得样品的PL 谱图及强度对比Fig.4 PL spectra and the peak intensity centered at 714 nm of samples prepared at different Ag contents
Ag2S-CdS 核壳结构的生成分两步进行,即先生成Ag2S 核,再引入Cd 源和S 源生长壳层。在第一步中,S 源的量对最终产物的光学性质的影响也是重要的。图5a 为加入Cd 前改变nAg∶nS比值所得样品的PL 发光谱。当nAg∶nS比为6∶2 时,在586 nm 附近出现一个宽的强发射峰,同时存在一个较弱的位于710 nm 的肩峰,当nAg∶nS比为6∶3 时,586 nm 峰逐渐消失,710 nm 峰显著增强,同时在666 nm 处出现一个新峰,继续增大比为6:4 时,两发射峰均增加到最强,随后逐渐下降,710 nm 峰稍许红移到718 nm 处。图5b 所示为生长Ag2S 时固定nAg∶nS=6∶4,生长壳层中改变nCd∶nS所得样品710 nm 发光峰强度随时间的变化关系。由图可知,随着S 源的不断增加,峰位基本不变,而强度持续增强,直到nCd∶nS为10∶6 时最大,随后下降。本实验中,从化学计量比角度看,S 前驱物量始终为不足,实验条件的改变会导致产物中Ag 和Cd 的相对含量不同,相关机理仍在进一步研究中。这些实验结果表明在核和壳的生长过程中改变S 源的使用量,也可有效调控量子点的发光峰位置和强度,同样可以实现近红外发光。
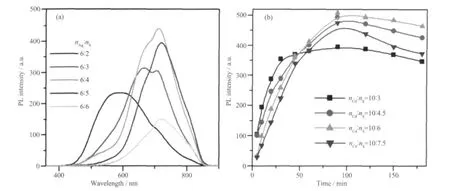
图5 (a) 生长核中改变S 源的量所得样品的PL 发光谱, (b) 生长Ag2S 时固定Ag∶S=6∶4,生长壳层中改变Cd∶S 所得样品710 nm 发光峰强度随时间的变化关系Fig.5 (a) PL spectra of sample when adding different S source during the growth of core; (b) PL peak intensities centered at 714 nm along with different Cd∶S ratios and reaction time during the shell growth
2.5 不同GSH 用量对光学性质的影响
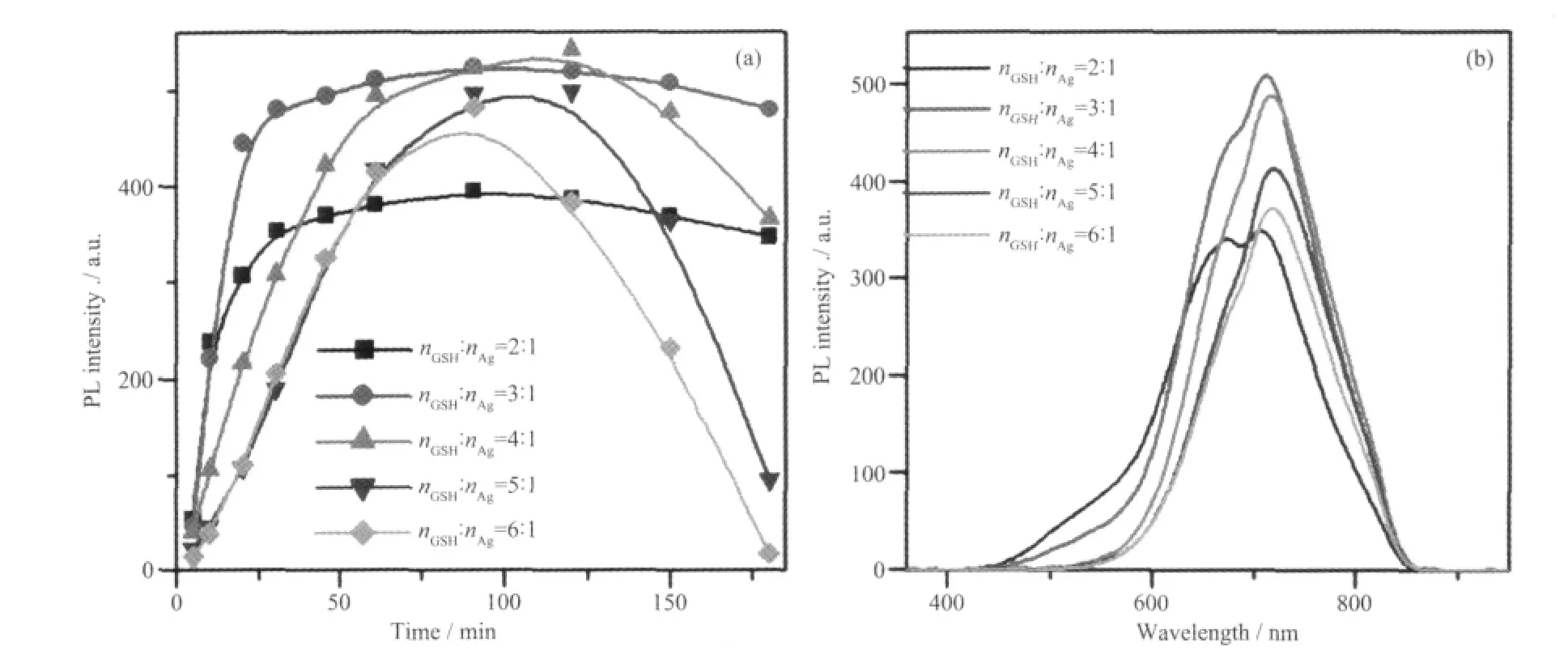
图6 GSH 用量不同所得量子点710 nm 处的发光强度变化(a)及PL 发光谱图(b)Fig.6 (a) PL peak intensities centered at 710 nm and (b) PL spectra when using different Ag contents
图6 为不同GSH 用量所得量子点710 nm 处的发光强度变化(a)及PL 发光谱图(b)。由图6b 可知,nGSH∶nAg为2∶1 时,在674 和710 nm 处出现2 个强度相近的发光峰,随GSH 量增加到3∶1 时,两峰位置几乎不变,强度均有增强,且近红外发光强度明显高于674 nm 处强度,其比值为115%。继续增加GSH 量,近红外发光相对强度进一步增加,直到最大值,且红移到716 nm,此后发射峰位置不再移动,但强度逐渐下降。GSH 为多肽,由3 个氨基酸组成,其中含2 个羧基、1 个巯基、1 个氨基及2 个亚氨基,这些基团除了为GSH 提供良好的水溶性以外,巯基、氨基和亚胺基均可以通过配位作用而吸附在量子点表面,从而增加了GSH 的配位能力。在本实验中,过量的GSH 作为表面活性剂,部分与金属离子结合后以(M-GSH)离子形式存在,部分以自由配体形式存在,GSH 的浓度将直接影响二者浓度的相对比值,进一步地影响量子点的光学性质[30-31]。当包裹剂用量过大时,会显著提高自由配体浓度而降低(M-GSH)离子的相对浓度,而量子点发光峰的位置和强度对(M-GSH)离子的相对浓度比较敏感,因此,使用适当含量的GSH 配体,可以更好地包裹在纳米晶体表面,钝化表面缺陷,调控量子点的荧光性质。
3 结 论
在水溶液中采用GSH 作为表面活性剂,通过先生成Ag2S 核,再于核外生长CdS 的两步方法,合成得到了高质量的Ag2S-CdS 核壳结构量子点,实现了710~718 nm 范围内的近红外发光,所得量子点稳定性好,具有长的荧光寿命,同时考察了GSH 的量,Ag 和S 源前驱物的含量对量子点光学性质的影响。实验结果表明,生长核过程中Ag 和S 源的多少同时影响量子点的发光位置和强度,量较少时不利于近红外发光,量过多时将使近红外发光强度下降,而GSH 量和壳层生长中S 源的量几乎只影响近红外发光强度,发光位置保持不变。这些实验条件对量子点光学性质的影响可能来源于量子点中组分的分布及表面缺陷的控制。所得水溶性量子点在生物成像及标识方面具有潜在的应用价值。
[1] ZHANG Pei-Gen(张培根), YU De-Cai(余德才), CHENG Chuan-Wei(程 传 伟), et al. Chinese J. Inorg. Chem.(Wuji Huaxue Xuebao), 2007,23(9):1662-1666
[2] Li Y F, Liu L L, FangX L, et al. Electronchim. Acta, 2012,65:1-6
[3] Li Y F, Bao J C, Han M, et al. Biosens. Bioelec., 2011,26(8):3531-3535
[4] Fang Z,Wu P,Zhong X H.Nanotechnology,2010,21:305604
[5] Pan Z X, Zhang H, Cheng K, et al. ACS Nano, 2012,6(5):3982-3991
[6] Liu Q, Han M, Bao J C, et al. Analyst, 2011,136(24):5197-5203
[7] Zhu Q S, Han M, Wang H S, et al. Analyst, 2010,135(10):2579-2584
[8] WANG Xue-Ting(王雪婷), YU Jun-Sheng(于俊生), XIE Ying(谢颖). Chinese J. Inorg. Chem.(Wuji Huaxue Xuebao),2007,23(7):1185-1193
[9] YANG Xu(杨旭), ZHOU Hong(周宏), SHEN Bin(沈彬),et al. Acta Phys.-Chim. Sin.(Wuli Huaxue Xuebao), 2010,1:244-248
[10]LEI Da(雷达), SHEN Yong-Tao(沈永涛), FENG Yi-Yu(冯奕钰), et al. Sci. China Tech. Sci.(Zhongguo Kexue: Jishu Kexue), 2012,55:903-912
[11]Smith, A M, Nie S M. Accounts Chem. Res., 2010,43:190-200
[12]Koenraad P M, Flatte M E. Nature Mater., 2011,10:91-100
[13]Santra P K, Kamat P V. J. Am. Chem. Soc., 2012,134:2508-2511
[14]Suyver J F, Wuister S F, Meijerink A. Phys. Chem. Chem.Phys., 2000,2:5445-5448
[15]DONG Yu-Ming(董玉明), WANG Guang-Li(王光丽), LI Zai-Jun( 李 在 均). Chinese J. Inorg. Chem. (Wuji Huaxue Xuebao), 2010,26(11):1981-1986
[16]Yousefi M H, Abdolhosseinzadeh A A, Fallah H R, et al.Modern Phys. Lett. B, 2010,24:2591-2599
[17]Shen Q H, Liu Y, Xu J, et al. Chem. Commun., 2010,46:5701-5703
[18]WANG Yi-Lin(王益林), WAN Xin(万鑫), LIU Sheng-Yan(刘 声 燕), et al. Chinese J. Inorg. Chem.(Wuji Huaxue Xuebao), 2012,28(1):97-102
[19]Robinson R D, Sadtler B, Demchenko D O, et al. Science,2007,317:355-358
[20]Zhu G X, Xu Z. J. Am. Chem. Soc., 2011,133:148-157
[21]Peng P, Sadtler B, Alivisatos A P, et al. J. Phys. Chem. C,2010,114:5879-5885
[22]Demchenko D O, Robinson R D, Sadtler B. ACS Nano,2008,2:627-636
[23]Ekimov A I, Hache F, Efros A L, et al. J. Opt. Soc. Am. B,1993,10:100-104
[24]Son D H, Hughes S M, Yin Y, Alivisatos A P. Science,2004,306:1009-1012
[25]Dalpian G M, Chelikowsky J R. Phys. Rev. Lett., 2006,96:226802
[26]Zou L, Fang Z, Gu Z, et al. J. Lumin., 2008,129:536-540
[27]Ichimura M, Goto F, Arai E. J. Appl. Phys., 1999,85:7411-7417
[28]Du F F, Zhang H, Du X L, et al. Mater. Chem. Phys., 2010,121:118-124
[29]Robinson R D, Sadtler B, Demchenko D O, et al. Science,2007,317:355-358
[30]Norman T J, Magana J D, Bridges F. J. Phys. Chem. B,2003,107:6309-6317
[31]Norris D J, Nan Y, Kennedy T A, et al. Nano Lett., 2001,1:3-7
