生物合成二酮哌嗪类化合物的环二肽合酶研究进展
2013-08-14高菊芳
韩 伟,高菊芳
(上海师范大学生命与环境科学学院,上海200234)
环二肽(Cyclodipeptides)是二酮哌嗪衍生物(Diketopiperazines,DKPs)家族中的一类化合物,在最初多肽的化学合成中被作为副反应产物[1,2],也是微生物的次级代谢产物中比较庞大的一类化合物。DKPs骨架是稳定的六元环,有2个氢键给体和2个氢键受体,而氢键被认为是药物与受体相互作用的一种方式,是非常重要的药效基团[3,4]。DKPs具有广泛的生物活性和药理学活性[5,6],如抗菌、抗病毒、抗肿瘤以及类似免疫抑制剂的活性[7,8],同时有研究认为DKPs是潜在的血脑屏障跨膜载体[9]。
人们在很长一段时间里认为自然界中环二肽的DKPs骨架只由非核糖体肽合成酶(Nonribosomal peptide synthases,NRPSs)参 与 催 化 合 成[10,11]。NRPSs是一大类多功能酶,由多个模块(Module)按一定的空间顺序组合而成,每一个模块能独立地将一个氨基酸整合到肽链上。一个典型的模块由缩合(Condensation,C)结构域、腺苷酰化(Adenylation,A)结构域和肽酰载体蛋白(Peptidyl carrier protein,PCP)结构域组成。以硫酯的形式通过几个模块将游离的氨基酸形成肽键,从而延伸肽链的长度,最终在硫酯酶(Thioesterase,TE)结构域的作用下以水解或者环化的方式将肽链解离下来[12],形成非核糖体肽类化合物。
之后,人们发现环二肽合酶(Cyclodipeptide synthases,CDPSs)也能参与催化DKPs类化合物的合成。这类酶能够直接跳过在NRPSs途径中A结构域活化氨基酸的步骤,利用现成的氨酰-tRNA(aa-tRNA)而不是游离的氨基酸作为底物来形成环二肽类化合物。近几年的研究揭示了CDPSs的晶体构型和独特的催化机制,作者在此就近年来CDPSs的研究进展进行综述。
1 CDPSs——新发现的催化合成环二肽骨架的一类酶
首先被发现的CDPS是2002年Sylvie等报道的诺尔斯氏链霉菌(Streptomyces noursei)中的albC基因,其编码的AlbC蛋白是一个非常小的酶,能够催化合成白诺氏菌素(Albonoursin,图1a)的骨架结构1-cyclo(α,β-dehydroPhe-α,β-dehydroLeu)(cFL),但 是与NRPSs几乎没有同源性,并且与任何已知的功能性蛋白均没有相关性[13,14]。与NRPSs催化合成环二肽的方式不同,CDPSs利用aa-tRNA而不是游离状态的氨基酸作为底物。随着研究的深入,Gondry等[13]确定其为一类新的参与催化合成DKPs结构的酶并被命名为环二肽合酶(CDPS)。

图1 白诺氏菌素(a)、Mycocyclosin(b)、普切明(c)的结构Fig.1 Structures of albonoursin(a),mycocyclosin(b),pulcherrimin(c)
此后报道了2个合成DKPs结构的CDPSs:结核分枝 杆 菌 (Mycobacterium tuberculosis)菌 株 中 的rv2275基因编码的Rv2275蛋白,催化合成Mycocyclosin(图1b)的骨架结构cyclo(L-Tyr-L-Tyr)(cYY)[15];地衣芽孢杆菌(Bacillus licheniformis)菌株中的 YvmCBlic蛋白,催化合成普切明(Pulcherrimin,图1c)的骨架结构 Cyclodileucine(cLL)[16]。通过对其晶体结构以及生物信息学进行分析,确定这2个蛋白属于CDPSs家族。对AlbC、Rv2275、YvmC-Blic以及一些潜在的CDPSs蛋白进行多序列比对(图2),发现这些CDPSs的同源性较低,仅有20%~26%的相似性[16]。结合突变实验,Gondry等[13]和 Sauguet等[17]认为,2个特殊的连续序列GxSxxN(Ser是与氨基酸结合的活性位点,Asn处的氨基酸主要为Asn变为其它极性氨基酸如Thr,也具有活性)和YxxxExP与CDPSs的催化功能有关[13]。

图2 7个CDPSs序列比对Fig.2 Protein sequence alignments of seven CDPSs(The catalytic residues are shown by a black background)
对AlbC、Rv2275、YvmC-Blic三个蛋白进行生化实验发现,纯化后的CDPSs蛋白利用带电荷的tRNAs(Charged tRNAs)来合成非核糖体肽类的次级代谢产物——二酮哌嗪类化合物[8,13]。这与 NRPSs(需要以游离的氨基酸作为底物)催化二酮哌嗪类化合物的合成不同。
2 CDPSs是一类以aa-tRNA作为底物的酶
目前已知的在微生物中利用aa-tRNA作为底物来合成代谢产物的酶共有5类:
(1)谷氨酰还原酶,它能将Glu-tRNA上的谷氨酸解离下来作为合成卟啉(Porphyrin)的底物;(2)氨基酸-磷酸甘油酯合成酶,它能利用aa-tRNA使细胞膜上的磷脂氨酰化;(3)FemXAB家族蛋白,它们参与肽聚糖的生物合成;(4)L/F转移酶,它参与蛋白质的翻转作用[8,16];(5)环二肽合成酶。前4类酶参与细菌的初级代谢,且只能催化2个氨基酸形成1个肽键,而CDPSs与前4类酶几乎没有相似的保守序列,催化活性区域也不同,且参与次级代谢产物的合成,催化2个aa-tRNA形成含2个肽键的环二肽化合物[17]。
已经取得很好研究进展的3个CDPSs(AlbC、Rv2275、YvmC-Blic)的蛋白晶体结构与酪氨酸-tRNA合成酶(Tyr-tRS,Ⅰ型aa-tRSs)的催化结构域的结构非常相像,如AlbC和Rv2275的蛋白晶体结构与来源于詹氏甲烷球菌(Methanococcus jannaschii)的 TyrtRS、YvmC-Blic的蛋白晶体结构与来源于酵母的Tyr-tRS的构型就比较接近[15-17]。
氨基酸-tRNA 合成酶(aa-tRSs)催化氨基酸和tRNA形成aa-tRNA的过程分为两个步骤:首先在ATP存在下,活化氨基酸形成酶联氨基酸中间体;然后将酶联氨基酸中间体上的氨基酸转移到相对应的tRNA 上 形 成 aa-tRNA[18,19]。与 aa-tRSs不 同 的 是:CDPSs直接利用aa-tRNA作为底物从而催化形成二酮哌嗪类化合物。
3 CDPSs与Ⅰ型aa-tRSs结构上的异同
Ⅰ型aa-tRSs(Tyr-tRS和 Trp-tRS)的催化结构域中含有 Rossmann折叠(Rossmann fold)和 CP1(Connective-polypeptide)[18,20],在已经明确晶体结构的3个CDPSs中发现类似的结构。3个CDPSs中也能找到与Ⅰ型aa-tRSs中氨基酸结合口袋结构相对应的活性氨基酸,由此可以认为CDPSs是HUP家族的一个新成员[21]。
但是Ⅰ型aa-tRSs与CDPSs蛋白的晶体结构有6个明显的区别:(1)Ⅰ型aa-tRSs中标志性的与ATP结合相关的模体如HIGH和KMSKS,在CDPSs中没有被发现,而且在Ⅰ型aa-tRSs中一些与ATP活化相关的氨基酸残基也不存在于CDPSs中,而是替换成其它的氨基酸,这表明CDPSs不能利用ATP来活化氨基酸生成aa-tRNA[17]。(2)Ⅰ型aa-tRSs是以同源二聚体的形式将2个活性位点交错结合在二聚体的表面,每个单体与一个aa-tRNA交联。而CDPSs的寡聚化状态与其不同,AlbC和YvmC-Blic是以单体(溶液中的晶体分析)形式存在,可能是它们与aa-tRNA结合后导致的二聚化(Ⅰ型aa-tRSs就是一类功能化二聚体,虽然在溶液中能得到它的二聚体),但是在CDPSs的催化结构域中疏水表面的氨基酸残基(它们能够帮助二聚体的形成)并不保守。Sauguet等[17]对AlbC进行二聚化模拟,发现二聚化后的AlbC会发生催化口袋的物理分离,从而导致不能形成双肽键。Rv2275虽然以同源二聚体的形式存在,但是其与二聚化相关的氨基酸残基在CDPSs中不是保守氨基酸,而且其催化口袋和活性氨基酸并不在二聚体的表面,而是分离的,而Ⅰ型aa-tRSs的活性位点存在于其双结构域的表面。Rv2275中具有明显的重排结构,能够阻止其与ATP的结合[22]。(3)已知的CDPSs晶体数据表明它们的催化口袋开口非常窄,游离的氨基酸或aatRNA不能够自由通过,这也与Ⅰ型aa-tRSs不同。推测CDPSs的催化口袋处于一种关闭的状态,在有tRNA与其结合后才能打开,从而实现其催化作用[16]。(4)CDPSs的CP1区域有一个很长的Loop结构[16]。(5)CDPSs中有一段很长的富含带正电氨基酸的序列,而Ⅰ型aa-tRSs没有这种现象。通过广泛的基因突变研究发现,AlbC中的碱性碱基对于环二肽的合成非常重要。YvmC-Blic中碱基的等电替换能够使其具备与tRNA结合的活性。从而推测这些带正电的碱性氨基酸可能与tRNA结合有关[8]。(6)CDPSs有一些新的活性位点,如Ser位点,研究表明Phe与AlbC的Ser37形成共价键,Rv2275的对应的Ser88被证实是结合氨基酸的活性位点[15]。此外还有研究表明,YvmC-Blic中的Tyr180和Glu184与识别LeutRNALeu的亮氨酸基团有关,在Rv2275以及YvmCBlic催化口袋入口处的几个保守氨基酸可能与其催化合成环二肽的机制有关,Rv2275催化口袋上的Asn251能够识别底物上酪氨酸部分的羟基基团,在YvmC-Blic中替换为Leu202后与底物中亮氨酸部分的识别有关[16]。
通过CDPSs和Ⅰ型aa-tRSs结构异同的比较可以较好地解释CDPSs为什么能利用aa-tRNA而不是游离的氨基酸作为底物来形成肽键的原因。
4 CDPSs的催化机制(图3)
CDPSs与aa-tRSs和氨基酸结合部分结构比较相像,催化形成肽键的机制有一定的相似性。已有的研究数据表明CDPS可能的催化机制是:利用2分子的aa-tRNA作为底物,通过一个连续的乒乓反应(Pingpong reaction)合成环二肽。其催化步骤分为三步:首先CDPSs位于酶催化口袋上的Ser(AlbC中是Ser37)的羟基基团作为亲核试剂进攻第一个aa-tRNA,并在 Tyr和 Glu(AlbC中分别是 Tyr178和Glu182)的矫正下(与氨酰基团以氢键结合)使氨基酸部分结合到CDPSs上而tRNA分离,形成一个氨酰-CDPSs共价中间体;然后氨酰-CDPSs共价中间体的氨基酸基团进攻结合到CDPSs上的第二个aa-tRNA的酯键,tRNA分离,形成二肽-CDPSs或者二肽-tRNA中间体;最后,二肽-CDPSs或者二肽-tRNA中间体经过分子内催化形成最终的环二肽骨架(DKPs)结构[8,16],这与NRPSs蛋白催化合成环二肽类化合物的机制不同。目前还没有报道证实这两个推测的中间体的存在。
5 DKPs骨架形成后的后修饰
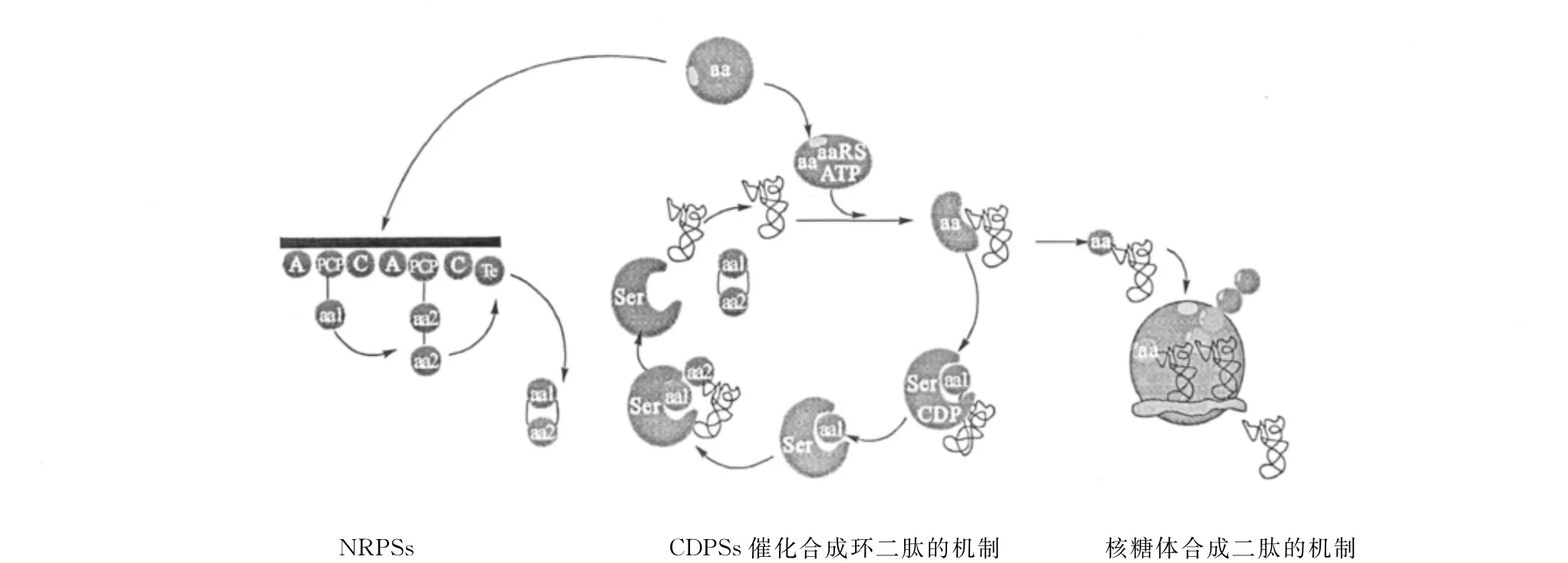
图3 CDPSs劫持aa-tRNA作为底物合成环二肽化合物和核糖体利用aa-tRNA合成核糖体肽的对比图Fig.3 The comparison of CDPSs hijack aa-tRNA to produce cyclodipeptides and the ribosome use aa-tRNA for the synthesis of peptide bonds
环二肽类化合物的骨架DKPs形成后,还需要一些后修饰才能形成结构复杂的环二肽化合物[23]。对比已知的CDPSs与NRPSs参与的环二肽形成,可以发现CDPSs催化合成DKPs中间体[13],然后在各种后修饰酶(大部分为氧化酶)的作用下,形成最终的环二肽化合物。目前通过基因组挖掘(Genome mining)发现与CDPSs基因簇共同起作用的后修饰的氧化酶有6类:细胞色素P450、FAD或NADH依赖的氧化酶、F420依赖的单加氧酶、α-酮戊二酸依赖的双加氧酶、铁氧化还原蛋白依赖的单加氧酶、α螺旋双铁氧化酶[21]。通过与CDPSs相连的后修饰酶的修饰作用,使得最终化合物的结构更丰富。目前已有3个编码CDPSs蛋白的基因被研究透彻,与它们相连的后修饰蛋白的后修饰作用没有共性,而是表现为3种不同的修饰形式:(1)细胞色素P450的CypX参与的二酮哌嗪环的氧化;参与Pulcherrimin形成的yvmC-cypX基因编码的CypX即CYP134A1蛋白是首个被发现的参与CDPSs生物合成的P450氧化酶[8]。它是血红素(Heme)依赖的单加氧酶,需要Fe3+作为外源性电子供体才能发挥其氧化酶活性,从而形成最终的化合物Pulcherrimin[16]。(2)Rv2275蛋白催化后的细胞色素P450参与的C-C芳基偶联;在Mycobacterium tuberculosis中发现的CYP121蛋白是研究比较透彻的环二肽后修饰酶,它与其它已知的P450氧化酶的功能不同,也参与产生菌株的脂代谢[24]。CYP121蛋白与Rv2275不仅在物理位置上是紧密相连的,而且共同参与了从Tyr-tRNA到Mycocyclosin的生物合成[8,25],CYP121蛋白使DKP环上的2个 Tyr侧链以C-C键偶联形成Mycocyclosin。(3)环二肽氧化酶(Cyclodipeptide oxidase,CDO)参与的α,β-脱氢反应,在很多微生物的次级代谢中α,β-脱氢氨基酸很常见[26]。CDO对许多环二肽的DKP环有活性,但是对线性二肽却没有作用,并且该酶对芳香族的氨基酸侧链有更好的活性。CDO在黄素作为辅因子的情况下,以O2作为电子供体同时产生副产物H2O2,催化环二肽的脱氢反应[8]。在大肠杆菌中AlbC和CDO共表达能够产生白诺氏菌素[14]。
6 CDPSs存在的广泛性
Belin等[8]运用生物信息学的方法,在50种不同细菌的基因组中找到可能编码CDPSs的基因,而且分布在不同的物种如放线菌、衣原体、真菌中,甚至在动物Platynereis dumerilii中也找到可能编码CDPSs的基因。他们通过对49个完整的可能CDPSs序列进行分析,最终确定了27个序列是可以被鉴定的,其中有18个是新的可以确定的CDPSs序列。但是就目前的数据来看,CDPSs并没有严格意义上的标志性蛋白序列,因此很难预测某个CDPSs催化形成其种产物,而且CDPSs蛋白的容忍性比较大,例如:AlbC在重组大肠杆菌中能够催化形成cFF、cFY、cFM、cYM、cYL、cLM、cLL、cMM 等多个化合物[8,17]。
7 NRPSs与CDPSs两种生物合成DKPs体系的对比
在自然界中,CDPSs和NRPSs体系都可以用来形成DKPs结构,但它们是有区别的:(1)NRPSs是一个多酶复合体,在合成DKPs时需要参与的蛋白很多,而CDPSs是一个非常小的酶(26kDa左右[8])。它们在大小上的不同是由于它们在形成肽键时采用的策略不同造成的:NRPSs需要A结构域识别、活化氨基酸并将其与T结构域以硫酯键连接,而CDPSs劫持现有的aa-tRNA参与合成化合物,没有氨基酸活化这个步骤。从而导致CDPSs只能利用20种结合在tRNA上的天然氨基酸作为底物来合成DKPs结构,而NRPSs识别的氨基酸就不局限于天然氨基酸,如利用邻氨 基 苯 甲 酸 来 合 成 Acetylaszonalenin[27]。 (2)NRPSs能够在与底物结合后对其进行修饰如甲基化,CDPSs途径只能在形成DKPs结构后通过后修饰酶的作用[21]形成最终化合物。因此,NRPSs途径合成的化合物的骨架具有更多的结构类型。
8 展望
虽然CDPSs可能的催化机制已被阐明,也通过生物信息学的方法找出了许多相关的蛋白,但是目前只有3个CDPSs蛋白被详细地阐明。与它们相连的后修饰酶也有3种不同的类型,因此,CDPSs体系相连的后修饰酶是否有共性、是否能找到类似NRPSs中甲基化等修饰作用的蛋白还需要进一步研究。Gondry等[13]对AlbC的改造证明了进行CDPSs改造的可行性,再加上CDPSs本身比较小的特性使其在酶工程中更容易改变和操作产生新的环二肽化合物。CDPSs是否从Ⅰ型aa-tRSs进化而来、改造Ⅰ型aa-tRSs能否形成CDPSs还需要进一步研究。随着新的CDPSs被发现和机制的阐明,更多的具有生物活性的DKPs化合物将会被发现。
[1]Giralt E,Eritja R,Pedroso E.Diketopiperazine formation in acetamido-and nitrobenzamido-bridged polymeric supports[J].Tetrahedron Letters,1981,22(38):3779-3782.
[2]Goodman M,Stueben K C.Peptide synthesis viaamino acid active esters.Ⅱ.Some abnormal reactions during peptide synthesis[J].Journal of the American Chemical Society,1962,84(7):1279-1283.
[3]Cui C B,Kakeya H,Osada H.Novel mammalian cell cycle inhibitors,spirotryprostatins A and B,produced by Aspergillus fumigatus,which inhibit mammalian cell cycle at G2/M phase[J].Tetrahedron,1996,52(39):12651-12666.
[4]Santamaría A,Cabezas N,Avendao C.Synthesis of tryptophandehydrobutyrine diketopiperazines and analogues[J].Tetrahedron,1999,55(4):1173-1186.
[5]Prasad C.Bioactive cyclic dipeptides[J].Peptides,1995,16(1):151-164.
[6]Huang R,Zhou X,Xu T,et al.Diketopiperazines from marine organisms[J].Chemistry & Biodiversity,2010,7(12):2809-2829.
[7]Bolognesi M L,Tran H N A,Ngoc H,et al.Discovery of a class of diketopiperazines as antiprion compounds[J].Chem Med Chem,2010,5(8):1324-1334.
[8]Belin P,Moutiez M,Lautru S,et al.The nonribosomal synthesis of diketopiperazines in tRNA-dependent cyclodipeptide synthase pathways[J].Natural Product Reports,2012,29(9):961-979.
[9]TeixidóM,Zurita E,Malakoutikhah M,et al.Diketopiperazines as a tool for the study of transport across the blood-brain barrier(BBB)and their potential use as BBB-shuttles[J].Journal of the American Chemical Society,2007,129(38):11802-11813.
[10]Sattely E S,Fischbach M A,Walsh C T.Total biosynthesis:In vitro reconstitution of polyketide and nonribosomal peptide pathways[J].Nat Prod Rep,2008,25(4):757-793.
[11]Strieker M,Tanovic'A,Marahiel M A.Nonribosomal peptide synthetases:Structures and dynamics[J].Current Opinion in Structural Biology,2010,20(2):234-240.
[12]Kopp F,Marahiel M A.Macrocyclization strategies in polyketide and nonribosomal peptide biosynthesis[J].Nat Prod Rep,2007,24(4):735-749.
[13]Gondry M,Sauguet L,Belin P,et al.Cyclodipeptide synthases are a family of tRNA-dependent peptide bond-forming enzymes[J].Nature Chemical Biology,2009,5(6):414-420.
[14]Lautru S,Gondry M,Genet R,et al.The albonoursin gene Cluster of S.noursei biosynthesis of diketopiperazine metabolites independent of nonribosomal peptide synthetases[J].Chemistry &Biology,2002,9(12):1355-1364.
[15]Vetting M W,Hegde S S,Blanchard J S.The structure and mechanism of the Mycobacterium tuberculosis cyclodityrosine synthetase[J].Nature Chemical Biology,2010,6(11):797-799.
[16]Bonnefond L,Arai T,Sakaguchi Y,et al.Structural basis for nonribosomal peptide synthesis by an aminoacyl-tRNA synthetase paralog[J].Proceedings of the National Academy of Sciences,2011,108(10):3912-3917.
[17]Sauguet L,Moutiez M,Li Y,et al.Cyclodipeptide synthases,a family of class-I aminoacyl-tRNA synthetase-like enzymes involved in non-ribosomal peptide synthesis[J].Nucleic Acids Research,2011,39(10):4475-4489.
[18]Ibba M,Sll D.Aminoacyl-tRNA synthesis[J].Annual Review of Biochemistry,2000,69:617-650.
[19]Woese C R,Olsen G J,Ibba M,et al.Aminoacyl-tRNA synthetases,the genetic code,and the evolutionary process[J].Microbiology and Molecular Biology Reviews,2000,64(1):202-236.
[20]Kuratani M,Sakai H,Takahashi M,et al.Crystal structures of tyrosyl-tRNA synthetases from Archaea[J].Journal of Molecular Biology,2006,355(3):395-408.
[21]Aravind L,de Souza R F,Iyer L M.Predicted class-I aminoacyl tRNA synthetase-like proteins in non-ribosomal peptide synthesis[J].Biology Direct,2010,5:48.
[22]Lahoud G,Hou Y-M.Biosynthesis:A new (old)way of hijacking tRNA[J].Nature Chemical Biology,2010,6(11):795-796.
[23]Gardiner D M,Cozijnsen A J,Wilson L M,et al.The sirodesmin biosynthetic gene cluster of the plant pathogenic fungus Leptosphaeria maculans[J].Molecular Microbiology,2004,53(5):1307-1318.
[24]Bordeaux M,Galarneau A,Fajula F,et al.A regioselective biocatalyst for alkane activation under mild conditions[J].Angewandte Chemie,2011,50(9):2075-2079.
[25]Belin P,Le Du M H,Fielding A,et al.Identification and structural basis of the reaction catalyzed by CYP121,an essential cytochrome P450in Mycobacterium tuberculosis[J].Proceedings of the National Academy of Sciences,2009,106(18):7426-7431.
[26]Gondry M,Lautru S,Fusai G,et al.Cyclic dipeptide oxidase from Streptomyces noursei[J].European Journal of Biochemistry,2001,268(6):1712-1721.
[27]Yin W B,Grundmann A,Cheng J,et al.Acetylaszonalenin biosynthesis in Neosartoryafischeri.Identification of the biosynthetic gene cluster by genomic mining and functional proof of the genes by biochemical investigation[J].Journal of Biological Chemistry,2009,284(1):100-109.
