Experimental and Molecular Dynamics Simulations for Investigating the Effect of Fatty Acid and Its Derivatives on Low Sulfur Diesel Lubricity
2013-07-31LuoHuiFanWeiyuLiYangZhaoPinhuiNanGuozhi
Luo Hui; Fan Weiyu; Li Yang; Zhao Pinhui; Nan Guozhi
(State Key Laboratory of Heavy Oil Processing, China University of Petroleum, Qingdao 266555)
Experimental and Molecular Dynamics Simulations for Investigating the Effect of Fatty Acid and Its Derivatives on Low Sulfur Diesel Lubricity
Luo Hui; Fan Weiyu; Li Yang; Zhao Pinhui; Nan Guozhi
(State Key Laboratory of Heavy Oil Processing, China University of Petroleum, Qingdao 266555)
In this work, fatty acid and its derivatives were adopted as lubricity additives for low sulfur diesel. Tribological evaluation obtained from the High-Frequency Reciprocating Rig (HFRR) apparatus showed that the lubricating performance of the additives increased in the following order: stearic acid>glycol monopalmitate>stearyl alcohol>ethyl palmitate>cetyl ethyl ether. The adsorption behavior of the additives on Fe (110) surface and Fe2O3(001) surface was investigated by molecular dynamics (MD) simulations to verify their lubricity performance. The results suggested that adsorption energies of the additives on Fe (110) surface are determined by the van der Waals forces, while adsorptions on Fe2O3(001) surface are significantly attributed to the electrostatic attractive forces. Higher values of adsorption energy of the additives on Fe2O3(001) surface indicate that the additive has more efficient lubricity enhancing properties.
MD simulation; adsorption; lubricity additive; fatty acid; low-sulfur diesel
1 Introduction
Due to environmental requirements, increasingly strict regulations on the sulfur content of diesel fuel were legislated, and petroleum refiners are engaging in production of low sulfur diesel with enthusiasm. However, the desulfurization process also removes from diesel fraction the oxygen and nitrogen compounds, which are responsible for the lubricity of diesel fuel[1-3]. Generally, when the sulfur content of petroleum-based diesel fuel is less than 500 μg/g, it could produce unacceptable wear[4]. Treating diesel fuel with lubricity additives have been proposed to compensate for the deterioration in natural lubricity of diesel and eliminate the excessive wear of rubbing surface. Oxygen containing compounds such as long chain fatty acids and their derivatives are superior lubricity additives because of their polarity-imparting oxygen atoms[5-9].
Boundary lubrication formed by the polar compounds plays a key role in the lubrication of fuels[10-11]. Lubricity additives have a high affinity to metallic surfaces and could form a thin protective metal-metal contact film[12-13]. This film was considered most important to reduce the number of points at which true metal to metal contact occurs. Rupture of the lubricant film will lead to deterioration in friction and wear[4], which suggests that the strength and stability of the film are key factors for diesel lubricity. An order of oxygenated lubricity additives (COOH>CHO>OH>COOCH3>C=O>C-O-C) has been obtained from studying various oxygenated 10-carbon-atom (C10) compounds[8]. In the case of the ethers, alcohols or ethyl acetate, alcohols appear to be better lubricants than ethers and ethyl acetate[3,6]. Although there have been several studies on the lubricity performance of oxygen compounds, the mechanism relating to the influence of molecular structure on the lubricity performance of the lubricity additive has rarely been studied.
The interactions between the additives and the metal surface are difficult to measure experimentally. However, applying the molecular dynamics (MD) simulation would be a powerful method to investigate the adsorption behavior at the microscopic molecular level and provide useful information[14-18]. Yanagisawa investigated the adsorptionof typical organic molecules onto Fe (110) surface using density functional theory[19]. The results showed that acetic acid, dimethyl ether, methylacetate and methanol did not generate appreciable chemical adsorption on the iron surface, and such adsorption is attributed to small physical interactions between a molecule and the iron surface, such as van der Waals and electrostatic interactions. These physical interactions could be obtained using molecular simulation method. Khaled used MD simulations to find the most stable adsorption sites for thiourea derivatives on Fe (110) surface[20]. It suggested that the efficiency order of thiourea derivatives adopted as corrosion inhibitor could be verified by the binding energies of these molecules adsorbed on iron surface. Therefore, the MD simulation could be used to describe the adsorption behavior of fatty acid and its derivatives on iron surface and provide further insight into their lubricity efficiency. The objective of this work is to investigate the lubricity evaluation of low sulfur diesel additized with fatty acid and its derivatives, such as alcohol, ester and ether, by a high frequency reciprocating rig (HFRR) apparatus. Subsequently, the adhesiveness of fatty acid and its derivatives on iron and iron oxide surfaces was examined by using the MD simulations, which could provide an interpretation of the correlations between the polar groups of lubricity additives and their lubricity enhancing performance.
2 Experimental Details and Computational Methods
2.1 Lubricity additive preparation
Five oxygen compounds, such as stearic acid (SA), stearyl alcohol (SAL), ethyl palmitate (EP), glycol monopalmitate (GMP) and cetyl ethyl ether (CEE) were chosen as lubricity additives. The esters were prepared via reaction of their corresponding fatty acid with alcohols in the presence of p-toluenesulfonic acid used as catalyst. The fatty acid and alcohols were mixed at a molar ratio of 1:1 and dissolved in toluene. The dosage of the catalysts was 1.0% of the reaction mixture. The reaction mixture was stirred at 80—160 ℃ for 5h. The reaction process was monitored through determination of acid value and hydroxy value of the product. Afterward, the reaction products were washed with 5.0% of aqueous sodium bicarbonate. The organic phase was dried over anhydrous sodium sulfate and then distilled in a vacuum evaporator to obtain the final products.
2.2 Lubricity measurements
A hydrotreated diesel supplied by the Qilu Petrochemical Company was adopted as the test fuel in this study, with its typical properties listed in Table 1. The sulfur and nitrogen content of the diesel was determined using an Antek 9000 sulfur and nitrogen fluorescence analyzer. Density, viscosity, and distillation range of the diesel sample were determined according to the standard method GB/T 2540—1981, GB 265—1975, and GB 255—1977, respectively. Additives were added to the test fuel in a concentration range from 50 μg/g to 500 μg/g. All lubricity measurements were performed by the High-Frequency Reciprocating Rig (HFRR) apparatus according to the ISO-12156 method.
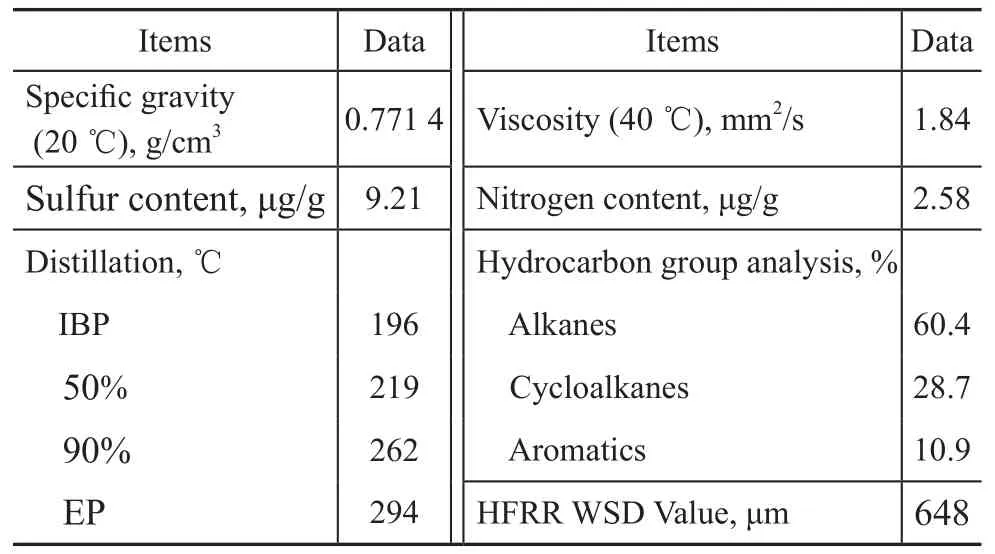
Table 1 Properties of the hydrotreated diesel fuel
2.3 MD simulations
During the desulfurization process, the polar compounds, such as nitrogen and oxygen compounds would be removed simultaneously, resulting in a diesel with low polar compounds content. It is difficult to form a protective layer on the metal surface for the low polar components. Therefore, the lubricity additives, such as SA, SAL, EP, GMP, or CEE, were adopted as adsorbate. Fe (110) surface and Fe2O3(001) surface were chosen as substrabte, and their sizes had dimensions of 3.475 3 nm×3.475 3 nm×1.418 8 nm, and 3.524 5 nm×3.524 5 nm×1.312 5 nm, respectively. The condensed-phase optimized molecular potentials for atomistic simulation studies (COMPASS)[21]forcefield as applied in Accelrys Materials Studio (MS) 5.5 was used for the MD simulations. The atom-based method with a cutoff of 1.55 nm was applied to computethe non-bonds Van der Waals and Coulomb interactions. All the energy minimization steps were performed by the Smart Minimizer algorithm, and the convergence criterion adopted for the value of maximum force was 102kcal/(mol·nm) for Steepest Descent, 1 kcal/(mol·nm) for Conjugate Gradient, and 10-6kcal/(mol·nm) for Newton-Raphson Gradient. The optimized models are shown in Figure 1.
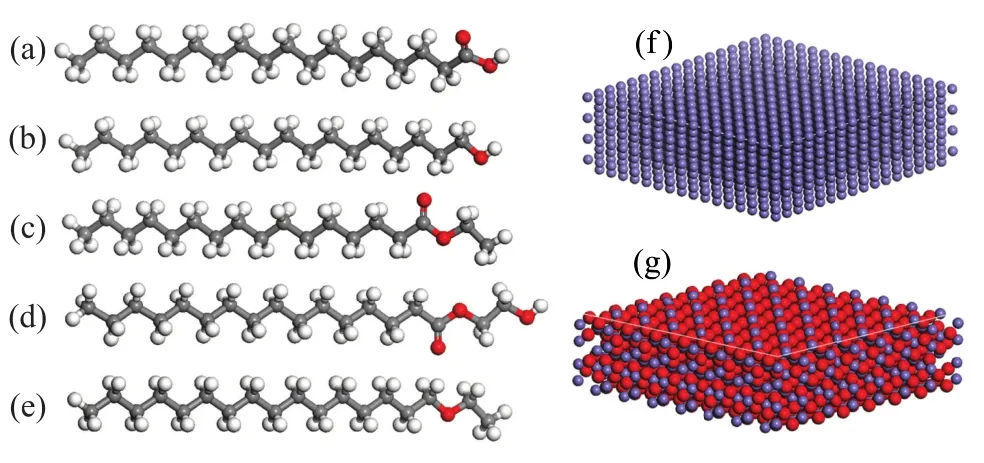
Figure 1 The optimized models
Adsorption Locator package included in MS software was utilized to identify preferential binding sites for an adsorbate molecule on the surfaces. Simulated annealing using the Metropolis Monte Carlo method has been implemented to calculate the adsorption density and the binding energy. Possible adsorption configurations have been sampled by carrying out Monte Carlo searches of the configuration space of the additives on the iron surface system as the temperature is gradually decreased. In this study, the automated temperature control was adopted, and 100 temperature cycles were employed for each run. The binding energy of adsorbates adsorbed on the surfaces is calculated as follows:
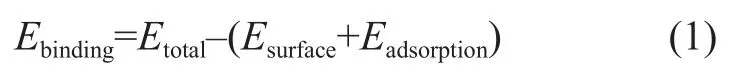
whereEtotalis the total energy of the surface and adsorbate,Esurfaceis the single-point energy of the surface, andEadsorbateis the single-point energy of adsorbate molecular.
3 Results and Discussion
3.1 Lubrication properties
The results of the HFRR analysis of fatty acid and its derivatives are shown in Figure 2. It can be seen from Figure 2 that the WSD of the base diesel fuel decreased regularly with an increasing concentration of these additives. There is a specific influence of the polar groups of the additives on their lubricating performance, and an order of enhancing lubricity (SA>GMP>SAL>EP>CEE) is obtained.
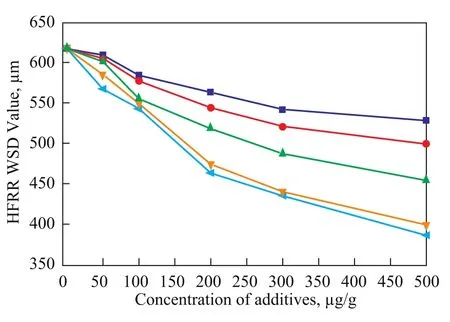
Figure 2 Effects of fatty acid and its derivatives on the lubrication properties of low-sulfur diesel fuel
3.2 The additives adsorbed on Fe (110) surface
Early studies have indicated that the adsorption forces of long-chain alkyl fatty acids, alcohols and esters on metal surfaces were largely physical[22], no evidence regarding the reaction of these organic compounds with zinc, iron, platinum, or gold was identified, although adsorption occurred. In the current study, to find out the preferential adsorption sites of the studied compounds on the iron surface and iron oxide surface, the adsorption behavior was investigated theoretically by MD simulation methods. The most suitable adsorption configurations for the oxygen compounds on Fe (110) surface are shown in Figure 3. It can be seen from Figure 3 that the oxygen-containing group and the carbon chain of the studied compounds remain almost parallel to Fe (110) surface. The adsorption density of the oxygen compounds on the Fe (110) surface is illustrated in Figure 4. High values of adsorption density indicate that the oxygen compounds are likely to adsorb on the iron surface and form stable films to protect iron from excessive wear.
The outputs and descriptors obtained from the adsorption locator module are listed in Table 2. The total energy (Etotal) of the substrate-adsorbate configuration is definedas the sum of the energies of the adsorbate components and the adsorption energy (Eadsorption). The substrate energy is taken as zero in adsorption locator module.Eadsorptionreports the energy released (or required) when the relaxed adsorbate component is adsorbed on the substrate and it is the sum of the rigid adsorption energy (ERigidadsorption) and the deformation energy (Edeformation) for the adsorbate components.ERigidadsorptionis defined as the energy released (or required) when the unrelaxed adsorbate components (i.e., before the geometry optimization step) are adsorbed on the substrate.EDeformationreports the energy released when the adsorbed adsorbate components are relaxed on the substrate surface. dEad/dNiis defined as the energy of substrate-adsorbate configurations where one of the adsorbate components has been removed. The binding energy (Ebinding) presented in Table 2 is calculated from Eq. (1), which consists of two parts: the van der Waals part and the electrostatic part.

Figure 3 The most suitable configuration of the additives adsorbed on Fe (110) surface

Figure 4 The adsorption density field of the additives adsorbed on Fe (110) surface
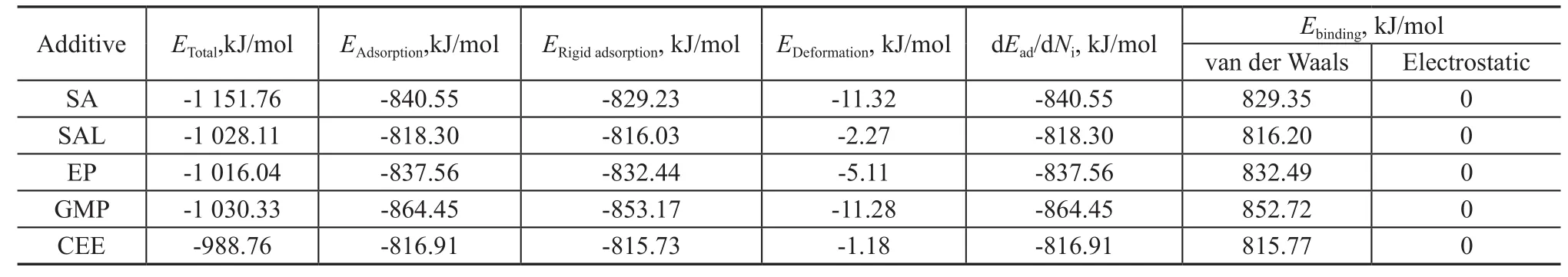
Table 2 Results obtained from the Mont Carlo simulation for adsorption of the additives on Fe (110) surface
As reported by Desai, et al.[23], the van der Waals interactions are dominant in adsorption of lone-pair electrons of oxygen on metal surfaces. The data listed in Table 2 also indicate that the binding energies between the oxygen compounds and Fe (110) surface are determined by the van der Waals forces while the electrostatic energies are zero. The binding energy and the adsorption energy of additives decrease in the following order: GMP>EP≈SA>SAL≈CEE, which can be attributed to the fact that the magnitude of van der Waals force depends on the relative molecular mass, and a high mass produces a larger force. In addition, the order of the binding energy is consistent with the case of adsorption density as shown in Figure 4.
3.3 The additives adsorbed on Fe2O3(001) surface
Because the clean iron surface is an uncharged surface, the interaction of the molecules with the iron surface isresulted from the van der Waals forces. There are no obvious effects of the functional groups on the adsorption intensity. Iron could react readily with oxygen to form a layer of oxide to protect the rest of the metal. The presence of this oxide layer on the iron surface could encourage the adsorption[24].
Figure 5 shows the most suitable adsorption configurations of the oxygen compounds on Fe2O3(001) surface obtained from MD simulations. The adsorption density of the molecules on the Fe2O3(001) surface is presented in Figure 6, and it is higher than the one on the Fe (110) surface, which suggests that the adsorbates have a higher affinity to iron oxide surfaces for forming a protective film.
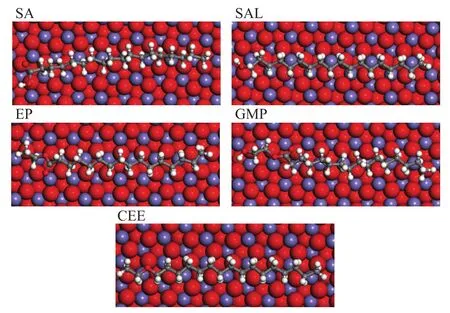
Figure 5 The most suitable configuration of the molecules adsorbed on Fe2O3(001) surface

Figure 6 The adsorption density field of the additives adsorbed on Fe2O3(001) surface
Actually, a past experimental study suggested that the extent of irreversible adsorption of polar organic compounds on steel was dependent on the functional groups and decreased in the following order: acids, alcohols, esters[25]. In particular, the value of adsorption density obtained from MD simulation decreases in the following order: SA>GMP≈SAL>EP>CEE, which is in good agreement with the experimental results. Therefore, the adsorption on the oxidized surface should be more prevalent for the oxygen compounds.
The outputs and descriptors of the adsorbates adsorbed on Fe2O3(001) surface are presented in Table 3. It can be seen from Table 3 that the binding energies of the oxygen compounds on Fe2O3(001) surface are much larger than in the case of Fe (110) surface, and the higher values of binding energy indicate that the presence of oxide layer on the iron surface could encourage the adsorption of the oxygen compounds on the surface, mainly via electrostatic attractive forces. There is little difference between the van der Waals forces of the molecules on Fe2O3(001) surface and the binding energies are originated mainly by the electrostatic attractive forces. Therefore, it is evidentthat ionic interactions of a metal substrate with a polar molecule due to hydrogen bonding and Debye orientation forces (electrostatic forces) are considerably stronger than those based on dipole (van der Waals) forces.
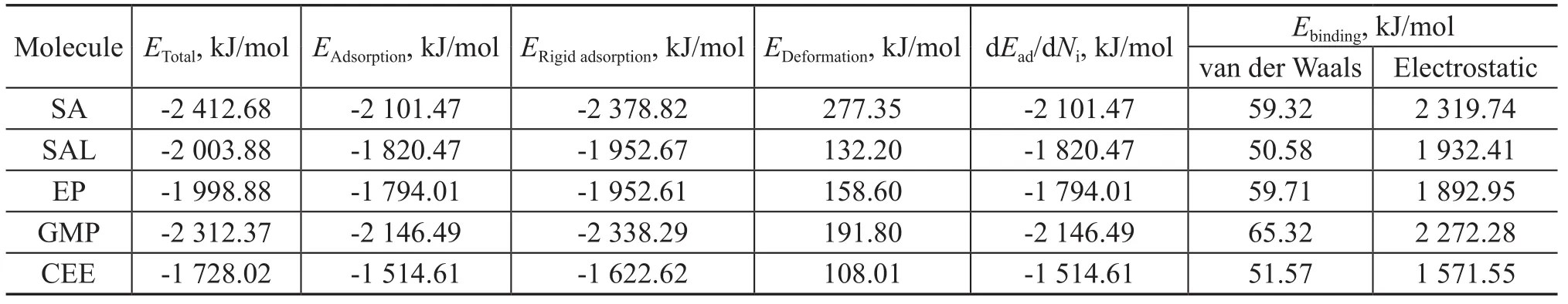
Table 3 Results obtained from the Mont Carlo simulation for adsorption of the additives on Fe2O3(001) surface
The data listed in Table 3 show that the binding energy of the adsorbates adsorbed on Fe2O3(001) surface decreases in the following order SA>GMP>SAL>EP>CEE. Judging from the polarity of the additives, it is suggested that the binding energy is confirmed to increase markedly with the increase in the polarity of functional groups. Further, the lubricity efficiency of additives is found to follow the same order of their binding energy on Fe2O3(001) surface. This means that the additive with a more polarity would have a higher affinity to the iron oxide surface and could improve the lubricity more effectively.
4 Conclusions
Fatty acid and its derivatives were adopted as lubricity additives for low sulfur diesel in this work. The results obtained from the HFRR lubricity evaluation showed an order of enhancing lubricity: SA>GMP>SAL>EP>CEE. Their lubricating performance was explained in terms of MD simulations.
The adsorption behavior of the additives on the surfaces obtained from MD simulations has demonstrated that binding energies of the additives on Fe (110) surface are determined by the van der Waals forces, while the adsorption of the additives on Fe2O3(001) surface is significantly attributed to the contribution of electrostatic attractive forces. The binding energies of the additives on Fe2O3(001) surface increase in the following order: SA>GMP>SAL>EP>CEE, which is consistent with the lubricity efficiency of the studied compounds obtained from the experimental measure. Higher values of the adsorption energy of the additive on the iron oxide surface indicate that the additive has more efficient lubricity enhancing properties.
Acknowledgment:This work was financially supported by“the Fundamental Research Funds for the Central Universities, China” (11CX06036A).
[1] Margaroni D. Fuel lubricity[J]. Ind Lubr Trib, 1998, 50 (3): 108-118
[2] Barbour R, Rickeard D, Elliott N. Understanding Diesel Lubricity[R]. SAE Paper 2000
[3] Anastopoulos G, Lois E, Zannikos F, et al. Influence of acetoacetic esters and di-carboxylic acid esters on diesel fuel lubricity[J]. Tribol Int, 2001, 34(11): 749-755
[4] Lacey P, Westbrook S. Diesel fuel lubricity[R]. SAE Paper 950248, 1995
[5] Anastopoulos G, Lois E, Serdari A, at al. Lubrication properties of low-sulfur diesel fuels in the presence of specific types of fatty acid derivatives[J]. Energy Fuels, 2001, 15(1): 106-112
[6] Anastopoulos G, Lois E, Zannikos F, et al. The tribological behavior of alkyl ethers and alcohols in low sulfur automotive diesel[J]. Fuel, 2002, 81(8): 1017-1024
[7] Geller D P, Goodrum J W. Effects of specific fatty acid methyl esters on diesel fuel lubricity[J]. Fuel, 2004, 83 (17/18): 2351-2356
[8] Knothe G, Steidley K R. Lubricity of components of biodiesel and petrodiesel: The origin of biodiesel lubricity[J]. Energy Fuels, 2005, 19 (3): 1192-1200
[9] Muñoz M, Moreno F, Monné C, et al. Biodiesel improves lubricity of new low sulphur diesel fuels[J]. Renewable Energy, 2011, 36 (11): 2918-2924
[10] Spikes H A, Cameron A. A comparison of adsorption and boundary lubricant failure[J]. Proc R Soc Lond A, 1974, 336: 407-419
[11] Belter M, Jahanmir S. Role of dispersion interactions between hydrocarbon chains in boundary lubrication [J]. Transaction, 1987, 30(11): 47-54
[12] Allen C M, Drauglis E. Boundary layer lubrication: monolayer or multilayer[J]. Wear, 1969, 14 (5): 363-384
[13] Hsu S M, Gates R S. Boundary lubricating films: formation and lubrication mechanism[J]. Tribol Int, 2005, 38(3): 305-312.
[14] Prathab B, Subramanian V, Aminabhavi T M. Molecular dynamics simulations to investigate polymer-polymer and polymer-metal oxide interactions[J]. Polymer, 2007, 48(1): 409-416
[15] Ylikantola A, Linnanto J, Knuutinen J, et al. Molecular modeling studies of interactions between styrene–butadiene latex and sodium polyacrylate polymer surface[J]. J Mol Struct (THEOCHEM), 2010, 953(1/3): 123-133
[16] Szefczyk B, Franco R, Gomes J A N F, et al. Structure of the interface between water and self-assembled monolayers of neutral, anionic and cationic alkane thiols[J]. J Mol Struct (THEOCHEM), 2010, 946(1/3): 83-87
[17] Awad M K, Mustafa M R, Abo Elnga M M. Computational simulation of the molecular structure of some triazolesas inhibitors for the corrosion of metal surface[J]. J Mol Struct (THEOCHEM), 2010, 959(1-3): 66-74
[18] Argyris D, Ho T, Cole D R, et al. Molecular dynamics studies of interfacial water at the alumina surface [J]. J Phys Chem C, 2011, 115(5): 2038-2046
[19] Yanagisawa S, Tsuneda T, Hirao K, et al. Theoretical investigation of adsorption of organic molecules onto Fe(110) surface[J]. J Mol Struct (THEOCHEM), 2005, 716(1-3): 45-60
[20] Khaled K F. Experimental, density function theory calculations and molecular dynamics simulations to investigate the adsorption of some thiourea derivatives on iron surface in nitric acid solutions[J]. Applied Surface Science, 2010, 256 (22): 6753-6763
[21] Sun H. COMPASS: anab initioforce-field optimized for condensed-phase applications-overview with details on alkane and benzene compounds[J]. J Phys Chem B, 1998, 102(38): 7338-7364
[22] Hackerman N, Roebuck A H. Adsorption of polar organic compounds on steel[J]. Ind Eng Chem, 1954, 46(7): 1481-1485
[23] Desai S K, Pallassana V, Neurock M. A periodic density functional theory analysis of the effect of water molecules on deprotonation of acetic acid over Pd (111)[J]. J Phys Chem B, 2001, 105(38): 9171-9182
[24] Smith H A, Allen K A. The adsorption ofn-nonadecanoic acid on metal surfaces[J]. J Phys Chem, 1954, 58(6): 449-452
[25] Cook E L, Hackerman N. Adsorption of polar organic compounds on steel[J]. J Phys Colloid Chem, 1951, 55(4): 549-557
Recieved date: 2013-04-15; Accepted date: 2013-05-04.
Prof. Fan Weiyu, E-mail: fanwyu@ upc.edu.cn.
杂志排行

中国炼油与石油化工的其它文章
- Study on the Synthesis and Properties of PET Using Hydrotalcite as Catalyst
- Kinetic Modeling of Methanol to Olefins (MTO) Process on SAPO-34 Catalyst
- CFD Simulation of Orifice Flow in Orifice-type Liquid Distributor
- Photocatalytic Denitrogenation over Modified Waste FCC Catalyst
- Synthesis of MgO-Al2O3/ZSM-5 by Solid State Reaction for Propane Dehydrogenation
- Study on Reactive Adsorption Desulfurization of Model Gasoline on Ni/ZnO-HY Adsorbent