气相色谱法测定鸡精中2种氯丙醇的方法研究
2013-07-28季晓娟王丹丹
黄 琦,季晓娟,王丹丹
(1.浙江科技学院 生物与化学工程学院,杭州310023;2.浙江省食品药品检验研究院,杭州310004)
氯丙醇是国际公认的食品污染物,氯丙醇的污染主要来源于酸水解植物蛋白(HVP),而HVP被广泛应用于各种调味品。蛋白水解物中氯丙醇的种类有:3-氯-1,2-丙二醇(3-MCPD)、2-氯-1,3-丙二醇(2-MCPD)、1,3-二氯-2-丙醇(1,3-DCP)和2,3-二氯-1-丙醇(2,3-DCP)。其中3-MCPD和1,3-DCP是主要检测的对象。毒理学研究表明3-MCPD有致癌性、遗传毒性、生殖毒性、神经毒性和免疫毒性等,是氯丙醇中的主要成分[1]。WHO/FAO食品添加剂联合专家委员会(JECFA)提出,3-MCPD的暂定每日最大耐受摄入量(PMTDI)为2μg(以每千克体质量计),认为1,3-DCP为遗传毒性致癌物,目前不宜制订每日耐受量(TDI)[2]。许多国家已经制订了HVP和酱油中3-MCPD的限量标准,欧盟要求HVP和酱油中的3-MCPD低于20μg/kg,德国和澳大利亚规定1,3-DCP应低于50μg/kg[3]。目前,对氯丙醇的研究主要集中在日常生活中用到的酱油[4-6],而鸡精作为常用调味品之一报道较少。氯丙醇定量的主要方法是气相色谱-质谱联用法,该法灵敏度高,专属性好,但是需要采用同位素内标定量,操作繁琐,成本较高,而且仪器价格昂贵,难以普及。本试验采用基质固相分散萃取法(MSPD)提取、净化样品,用七氟丁酰基咪唑(HFBI)衍生化,建立气相色谱-电子捕获检测器(GC-ECD)测定鸡精中3-MCPD和1,3-DCP的方法。实验表明,该方法简便易行,灵敏度高,结果令人满意,具有较强的实用性。
1 材料与仪器
3-MCPD和1,3-DCP(纯度≥99.0%):德国 Merck公司,HFBI:美国Pierce公司;ExtrelutTM20硅藻土填料:德国Merck公司;正己烷(色谱纯):美国Sigma公司;乙醚为分析纯,重蒸后使用;氯化钠和无水硫酸钠为分析纯,经过600℃灼烧4h后得到;水为超纯水;鸡精购于本地超市;除非另有说明,实验所用试剂均为分析纯。
岛津GC-14C型气相色谱仪配电子捕获检测器(ECD):日本岛津公司,色谱柱:J&WDB-5(30m×0.32mm×0.25μm),玻璃层析柱(40cm×2cm),旋转蒸发器,氮气蒸发器,恒温箱,涡漩混合器,1mL气密针。
2 试验部分
2.1 样品的提取、净化
称取试样2.00g,置100mL烧杯中,加饱和氯化钠溶液(称取氯化钠290g,加水溶解并稀释至1 000mL)6g,超声15min。加入5g ExtrelutTM20硅藻土柱填料,搅拌均匀后,装入已依次充填1cm高的无水硫酸钠和5g ExtrelutTM20的玻璃层析柱中,压实,再填入1cm高的无水硫酸钠,放置15min后,用正己烷洗脱去除脂溶性杂质,并弃去。用乙醚140mL洗脱,流速约5mL/min,收集乙醚洗脱液。在收集的乙醚洗脱液中加无水硫酸钠15g,振摇,放置10min后,过滤,滤液于40℃温度下旋转蒸发至约0.5mL(切勿蒸干),转移至约5mL具塞试管中,用正己烷洗涤旋转蒸发瓶,合并洗涤液至试管中,并用正己烷稀释至刻度。
2.2 衍生化
将上述试样提取液在室温下用氮气浓缩至2.0mL,用气密针迅速加入HFBI 50μL,立即密塞。涡旋充分混合后,于75℃下保温30min。取出后,放置至室温,加饱和氯化钠溶液3mL;涡旋混合0.5min,静置使两相分离。取上层正己烷加无水硫酸钠约0.3g干燥,静置5min,供气相色谱分析测定[3]。
2.3 标准溶液的配置
分别准确称取3-MCPD和1,3-DCP标准品25mg(有效成分),分别置于2个25mL容量瓶中,用乙酸乙酯溶解并定容,得质量浓度为1000mg/L的标准储备液。用正己烷稀释10倍,配制100mg/L中间溶液,最后用正己烷配成质量浓度为0.10、0.50、1.00、1.50、2.00、2.50mg/L的混合标准溶液系列。取上述混合标准溶液系列各0.2mL,用正己烷定容至2.0mL,用气密针迅速加入HFBI 50μL,立即密塞,振匀。接下来的步骤与样品的衍生化方法相同,得到质量浓度范围在0.010~0.250mg/L的标准系列HFBI衍生物溶液。
2.4 气相色谱条件
DB-5石英毛细管柱,30m×0.32mm×0.25μm;进样口温度为240℃,检测器温度为280℃;色谱柱温度初温为60℃,保持1min,以4℃/min升至100℃,保持1min,再以30℃/min升至250℃,并保持5min;载气为高纯氮气,流速为2.0mL/min;进样方式为不分流进样,进样量1.0μL;峰面积外标法定量。
3 结果与讨论
3.1 样品提取净化条件的选择
样品纯化时,需要清除基质中的色素、脂肪和蛋白质,而氯丙醇在极性溶剂和非极性溶剂中都有良好的溶解性,所以液液提取方法很难有效进行,必须采用固相提取技术[7]。试验考察了氧化铝、硅胶、ExtrelutTM20硅藻土3种吸附剂对样品的吸附作用,结果表明,ExtrelutTM20硅藻土效果最佳,其稳定性好、重现性好,具有很好的吸水性。装柱时比较了2种方式:一是常规方式用ExtrelutTM20硅藻土装柱;二是把该ExtrelutTM20硅藻土一半先装柱,另一半与样品混合均匀后再装柱。试验结果表明,后者更加有利于提取操作。
采用有机溶剂预洗脱去除样品中的脂溶性杂质和色素等,可减少衍生时的干扰及对色谱柱进样口的污染。试验表明,仅测定3-MCPD时可用正己烷-乙醚(体积比9∶1)洗脱去除脂溶性杂质,但是1,3-DCP有较大的损失(≥30%),采用正己烷预洗脱即可避免损失。
洗脱氯丙醇常用乙醚或乙酸乙酯[7]。通过试验比较了这2种洗脱溶剂,回收率如表1所示。结果表明,采用乙酸乙酯洗脱,用量较大,且杂峰太多,而采用乙醚洗脱,效果较好。采用140mL乙醚洗脱,回收率可达98.2%以上。洗脱液含有水分会影响后续的衍生化,采用无水硫酸钠干燥,效果较好,而采用硫酸镁、硫酸钙、氧化钙等干燥会引起较大的损失。
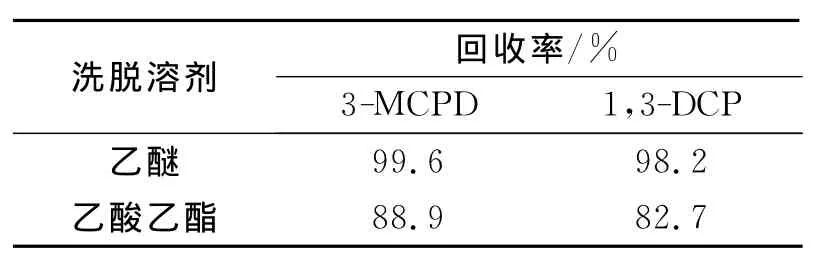
表1 2种洗脱溶剂的回收率(n=6)Table 1 Recoveries of 2elution solvents(n=6)
3.2 衍生化方法的选择
3.2.1 衍生化试剂的选择
氯丙醇的极性较大、蒸气压低、沸点较高,衍生化后可以提高分析物的挥发性及检测的灵敏度。可供使用的衍生化试剂有七氟丁酰试剂 (HFBI、HFBA)、硼酸类(丁基硼酸、苯基硼酸)、丙酮或庚酮、三氟乙酸酐(TFAA)、N,O-双三甲基-三氟乙酰胺(BSTFA)等试剂。硼酸类、丙酮只能衍生3-MCPD等双羟基醇类化合物;TFAA比HFBI的衍生反应容易进行,但产生的有机酸可能会分解氯丙醇衍生物,且灵敏度不如HFBI衍生物高;七氟丁酸酐(HFBA)虽然在室温下就可以反应,但是副产物七氟丁酸可能会降解氯丙醇衍生物;BSTFA主要用于氢火焰离子化检测器(FID)[8]。相比之下,采用HFBI衍生后氯丙醇的电负性明显增强,且HFBI衍生的化合物比较稳定、检测灵敏度较高。
3.2.2 衍生化条件的选择
分别选择60、70、80、90℃等不同的衍生化温度,该衍生化反应在70~80℃效果最佳,最终选择75℃为衍生化温度;在该温度下分别衍生20、30、40、50min,结果表明,衍生时间大于30min后,峰面积增加不明显,因此选择反应时间30min。在选定的衍生化时间及温度下,分别加入40、50、60、70μL的衍生化试剂,试验发现50μL的HFBI已完全满足要求。由于HFBI遇水迅速反应,因此环境的湿度影响极大,使用过程中需注意防潮。在相对湿度超过70%时,需用气密针迅速加入衍生化试剂。衍生后加入饱和氯化钠或蒸馏水除去过量衍生剂、副产物。
3.3 色谱条件的选择
衍生化后的氯丙醇可用非极性柱或弱极性柱分析。比较了实验室的2种色谱柱DB-1色谱柱(30m×0.32mm×0.25μm)和DB-5色谱柱(30m×0.32mm×0.25μm),均能满足分析要求;但是采用DB-5柱分析,峰形好,出峰快,因此选用DB-5色谱柱。为使杂质峰和主峰获得较好的分离效果和合适的保留时间,必须采用程序升温方式。经多次试验,所选条件同2.4。在该色谱条件下,3-MCPD和1,3-DCP的保留时间分别为约7.23min和11.85min。衍生化后的3-MCPD和1,3-DCP标准溶液色谱图见图1,含氯丙醇的鸡精样品色谱图见图2。
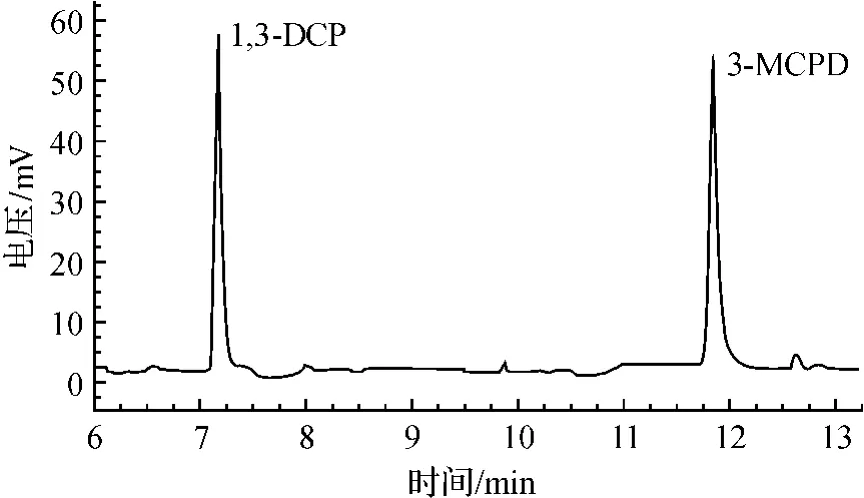
图1 3-MCPD和1,3-DCP标准HFBI衍生物气相色谱图Fig.1 Gas chromatogram of HFBI derivatives of 3-MCPD and 1,3-DCP standards
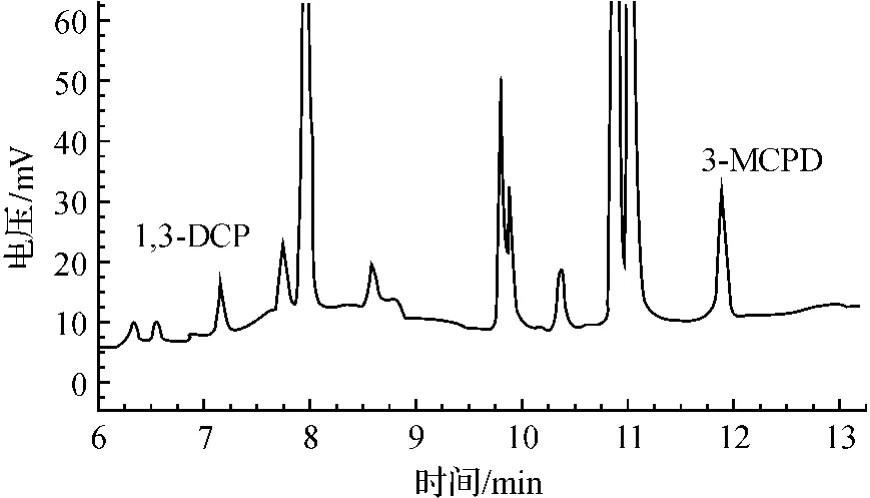
图2 鸡精样品中3-MCPD和1,3-DCP的HFBI衍生物气相色谱图Fig.2 Gas chromatogram of HFBI derivatives of 3-MCPD and 1,3-DCP in chicken essence
3.4 线性关系及检测限
分别取混合标准系列HFBI衍生物溶液1.0μL进样测定。分别以3-MCPD和1,3-DCP的质量浓度为横坐标,峰面积为纵坐标绘制标准曲线。3-MCPD的回归方程为:Y=1 136 457X-412.62,相关系数为0.999 1;1,3-DCP的回归方程为:Y=138 739X-337.15,相关系数为0.999 3。结果表明,2种氯丙醇在0.010~0.250mg/L范围内,线性关系良好。经多次试验,该方法测定3-MCPD和1,3-DCP的定量限均为0.01mg/kg,能满足检测和定量分析的要求。以3倍基线噪音计算,方法的检测限均为0.005mg/kg。
3.5 加标回收率试验和方法精密度
取3-MCPD和1,3-DCP空白的鸡精样品作为基质,分别测定0.05、0.10和0.20mg/kg 3种添标水平下3-MCPD和1,3-DCP的加标回收率,并计算相对标准偏差。每个水平测定6次,回收率及精密度结果见表2。结果表明,3-MCPD的回收率在95.7%~102.2%范围内;1,3-DCP的回收率在94.5%~99.3%范围内,两者相对标准偏差均小于5.0%。该方法具有较好的准确度和精密度,符合氯丙醇污染测定的技术要求。

表2 方法的回收率和精密度(n=6)Table 2 Recovery and precision of method(n=6)
3.6 鸡精样品检测
采用建立的试验方法对市场上12种品牌(每种品牌10批),共120份鸡精产品进行测定,共有4个品牌的21份鸡精样品中检出3-MCPD,检出率为17.5%,其中4份质量浓度超过20μg/kg;有3个品牌13份鸡精样品中检出1,3-DCP,检出率为10.8%,质量浓度均未超过20μg/kg。
4 结 语
本试验所建立的方法能同时测定鸡精中3-MCPD和1,3-DCP的质量浓度,方法灵敏度较高,回收率较高,相对偏差较小,可以准确地测定鸡精中2种氯丙醇的质量浓度;方法定量限0.01mg/kg,能满足鸡精中2种氯丙醇限量检测的要求;试验所用仪器易于普及,便于推广应用,是一种较理想的分析方法。本方法也适用于其他调味品和存在氯丙醇污染的食品中3-MCPD和1,3-DCP的限量测定。
[1]Baer I,De La Calle B,Taylor P.3-MCPD in food other than soy sauce or hydrolysed vegetable protein(HVP)[J].Analytical and Bioanalytical Chemistry,2010,396(1):443-456.
[2]FAO,WHO.Summary of the fifty-seventh meeting of the joint FAO/WHO expert committee on food additives[Z].Rome:FAO/WHO,2001:20-21.
[3]GB/T 5009.191—2006,食品中氯丙醇含量的测定[S].
[4]赵文杰,卢奎,冯业振.气相色谱-电子捕获检测法测定酱油中1,3-二氯-2-丙醇的含量[J].食品科技,2010,35(11):285-288.
[5]周相娟,谢精精,赵玉琪,等.气相色谱-质谱法测定酱油中氯丙醇类化合物[J].中国调味品,2011,36(5):88-90,120.
[6]Xu X M,Ren Y P,Wu P G,et al.The simultaneous separation and determination of chloropropanols in soy sauce and other flavoring with gas chromatography-mass spectrometry in negative chemical and electron impact ionization modes[J].Food Additives and Contaminants,2006,23(2):110-119.
[7]赵广西,杜伟,刘浩,等.酱油中氯丙醇测定的注意事项[J].中国调味品,2011,36(9):23-25.
[8]傅武胜,吴永宁.食品中氯丙醇测定方法研究进展[J].食品科学,2007,28(3):353-357.
