UPLC-MS/MS法测定猪肉中万古霉素与去甲万古霉素
2013-07-13冼燕萍陈立伟罗海英郭新东吴玉銮罗东辉侯向昶
冼燕萍,陈立伟,罗海英,郭新东,吴玉銮,罗东辉,侯向昶
(广州市质量监督检测研究院 国家加工食品质量监督检验中心(广州) 广州市食品安全检测技术重点实验室
广州市食品安全风险动态监测与预警研究中心,广东 广州 510110)
万古霉素和去甲万古霉素(结构式如图1所示)属于糖肽类抗生素,常用于治疗细菌感染。农业部第560号公告规定万古霉素为禁用兽药[1],如果这类药物被违规滥用于动物的养殖,很可能因其耐药性问题而影响动物性食品的安全、公共卫生以及出口贸易,长期食用则有可能导致消费者产生更严重的抗生素耐药性。万古霉素被列入卫生部发布的《食品中可能违法添加的非食用物质和易滥用的食品添加剂名单(第1-5批汇总)》,并注明“需要研制动物性食品中测定万古霉素的液相色谱-串联质谱法”[2]。因此,开展动物性食品中万古霉素等糖肽类抗生素残留量的检测技术研究迫在眉睫。
国内外关于万古霉素等糖肽类抗生素[3-14]的检测方法有高效液相色谱法[5-8]、高效液相色谱-质谱联用法[9-14],但检测对象多为血液样品[5-8],而关于食品中万古霉素的分析方法报道较少。刘佳佳等[10]采用液质联用法检测了牛奶中万古霉素的含量。本文建立了猪肉中万古霉素和去甲万古霉素的超高效液相色谱串联三重四极杆质谱(UPLC-MS/MS)检测方法,方法简便快速、灵敏度高、重现性好、实用性强,可为政府进一步打击违法添加非食用物质和推动食品安全标准体系的完善提供技术支持。
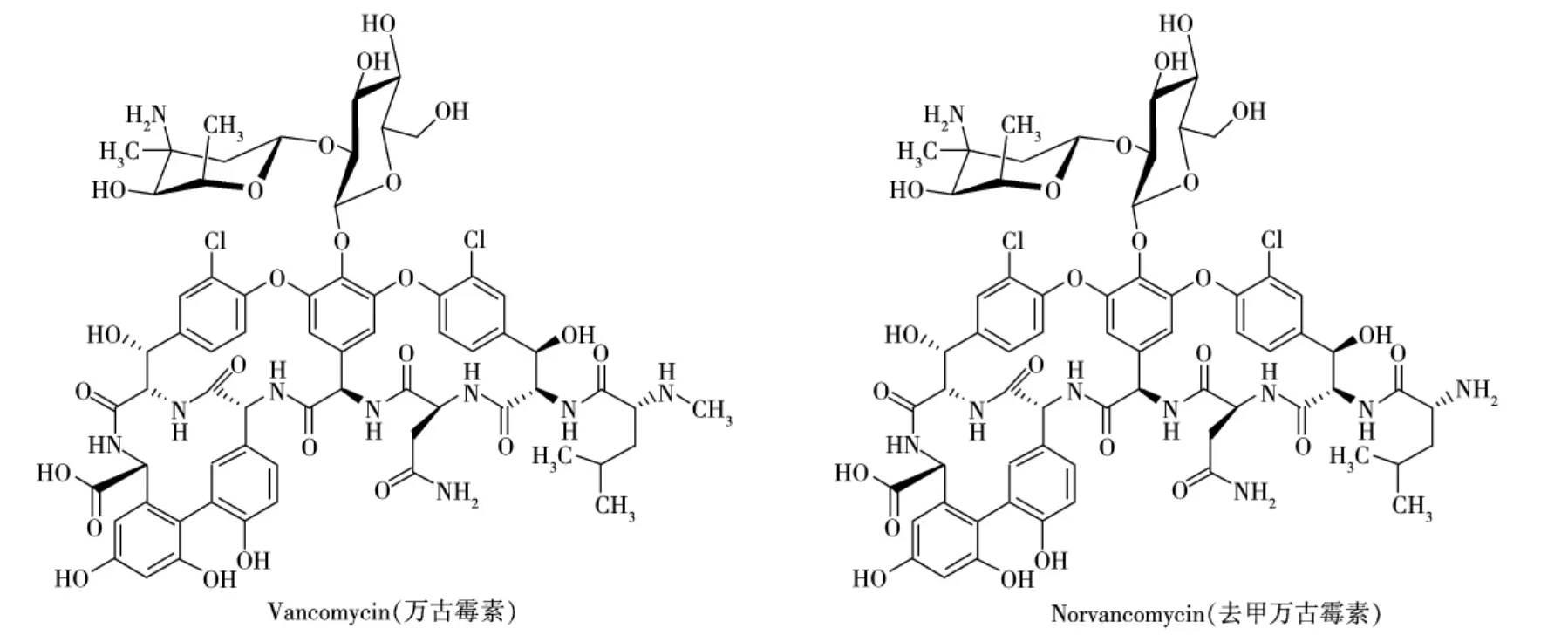
图1 万古霉素和去甲万古霉素的化学结构式Fig.1 Chemical structures of vancomycin and norvancomycin
1 实验部分
1.1 仪器与试剂
ACQUITYTM超高效液相色谱仪和Waters XevoTMTQ MS三重四极杆串联质谱仪(美国Waters公司);高速离心机5804R(德国Eppendorf公司);T18均质器、MS3 Basic漩涡混合器(德国IKA公司);KQ-250DV型数控超声波清洗仪(昆山市超声仪器有限公司);N-EVAP 112水浴氮吹仪(美国OA公司);Milli-Q去离子水发生器(美国Millipore公司);固相萃取装置(美国Waters公司);LC-C18固相萃取小柱(CNW公司,200 mg/3 mL):临用前依次用3 mL甲醇和3 mL水活化,3 mL 0.1%甲酸水-乙腈(9∶1,体积比)平衡。
万古霉素(含量95%,Sigma公司);去甲万古霉素(含量83.4%,中国药品生物制品检定所);甲醇和乙腈(HPLC级,德国Merck公司),甲酸(HPLC级,CNW公司),正己烷(分析纯,广州化学试剂厂),超纯水(18.2 MΩ)。10例猪肉样品均购于本地市场。
1.2 标准溶液的配制
分别准确称取万古霉素和去甲万古霉素标准品,用0.1%甲酸水溶液配成100 mg/L的单标标准储备液,4℃冰箱保存。
根据需要,用经前处理所得的阴性样品基质溶液配成不同浓度的系列基质匹配混合校准工作液,当天配制。以基质匹配校准工作曲线进行定量。
1.3 样品前处理
1.3.1 提 取 称取已均质的试样5.0 g(精确至0.001 g)于50 mL塑料离心管中,加入5 mL 0.1%甲酸水-乙腈(7∶3),涡漩3 min,超声10 min,4 000 r/min离心5 min,吸取上清液于10 mL比色管中,重复提取1次,合并上清液,定容至10 mL。取5 mL提取液于15 mL塑料离心管中,加入5 mL正己烷(乙腈饱和),涡漩2 min,12 000 r/min离心3 min,弃去正己烷层,重复用正己烷脱脂1次,下层提取液待净化。
1.3.2 净 化 吸取2.0 mL待净化液于试管中,加入4.0 mL 0.1%甲酸水,混匀,加入已活化的LC-C18固相萃取小柱中,控制流速不大于1 mL/min,待液体全部流出后,分别用2 mL 0.1%甲酸水-乙腈(9∶1)溶液和1 mL 50%甲醇润洗试管,转入小柱,弃去淋洗液,然后用8 mL甲醇洗脱,收集洗脱液,氮吹至近干,用0.1%甲酸水-乙腈(9∶1)溶解,转移、定容至1.0 mL,样液过0.22 μm滤膜后,供UPLC-MS/MS分析。
1.4 UPLC-MS/MS检测条件
色谱柱:Waters ACQUITY UPLC BEH C18(2.1 mm×100 mm,1.7 μm);流动相:A为0.1%甲酸水,B为乙腈,梯度洗脱程序:0.0~0.5 min,95%A;0.5~1.5 min,95%~85%A;1.5~3.0 min,85%~75%A;3.0~3.1 min,75%~95%A;3.1~5.0 min,95%A;流速:0.25 mL/min;柱温:30℃;进样量:5 μL。
质谱条件:ESI正模式;毛细管电压1.0 kV;离子源温度150℃;去溶剂气温度500℃;去溶剂气:氮气,流速800 L/h;锥孔气:氮气,流速50 L/h;碰撞气:高纯氩气,流速0.2 mL/min;检测模式:多反应监测(MRM)模式;2种化合物的监测离子对(m/z)、丰度比及其它优化的质谱参数(锥孔电压、碰撞能等)见表1,每个离子对的驻留时间均为0.1 s。

表1 目标化合物的质谱分析条件Table 1 MS parameters for the analysis of vancomycin and norvancomycin
2 结果与讨论
2.1 质谱条件的选择
万古霉素和去甲万古霉素均含有一个二氯三苯基醚结构单元、两个糖基和多个氨基酸,去甲万古霉素比万古霉素少1个甲基,两者的分子量分别为1 449和1 435。在一级质谱中,易与H+结合,产生带正电的双电荷离子[M+H]2+,质谱图上呈现m/z 725.6和m/z 718.1,以此作为万古霉素和去甲万古霉素的母离子,并优化锥孔电压,使母离子强度最大。带多电荷的母离子进入二级质谱后,发生断裂或重排等反应产生不同的碎片离子,优化碰撞电压,使每种化合物的主要特征碎片离子的强度达到最大,并以丰度最大的两个碎片离子作为定性与定量离子。2种目标物的子离子扫描质谱图如图2所示,特征离子对和电离条件、碰撞条件见表1。
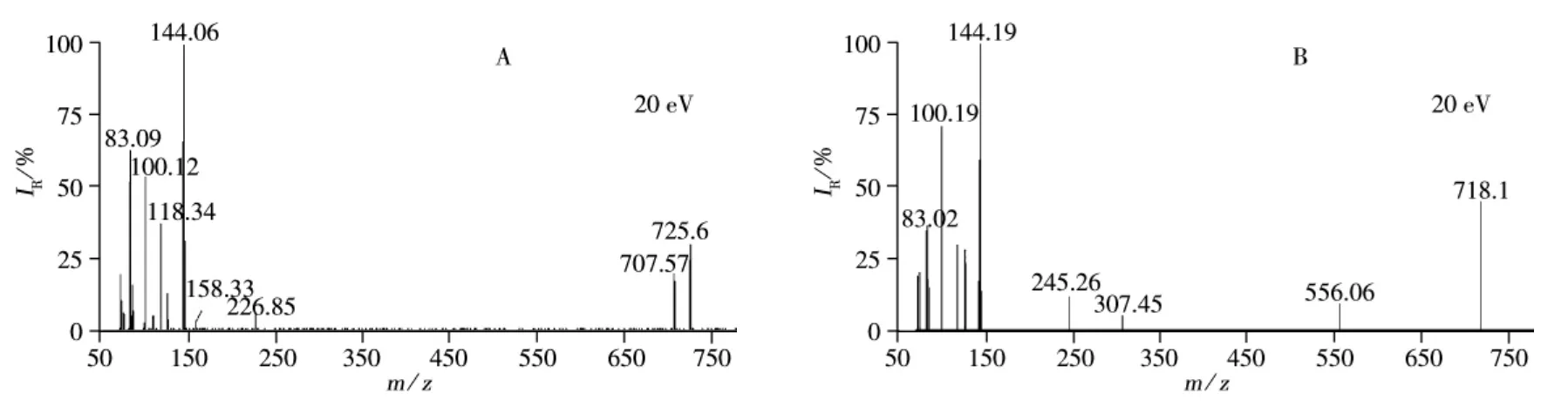
图2 万古霉素(A)和去甲万古霉素(B)的子离子扫描图Fig.2 Daughter scan spectra of vancomycin(A)and norvancomycin(B)
2.2 色谱条件的优化
在流动相中加入甲酸,可以为目标化合物提供必需的质子来源,提高离子化效率。用等度洗脱或梯度洗脱变化太快时,由于万古霉素和去甲万古霉素的结构仅相差1个甲基,二者不能完全分离。采用“1.4”所示的梯度洗脱,可将2种目标物分离,万古霉素和去甲万古霉素的保留时间分别为2.72 min和2.64 min,目标物混合标准溶液(5.0 μg/L)的总离子流图(TIC)和提取离子色谱图见图3。
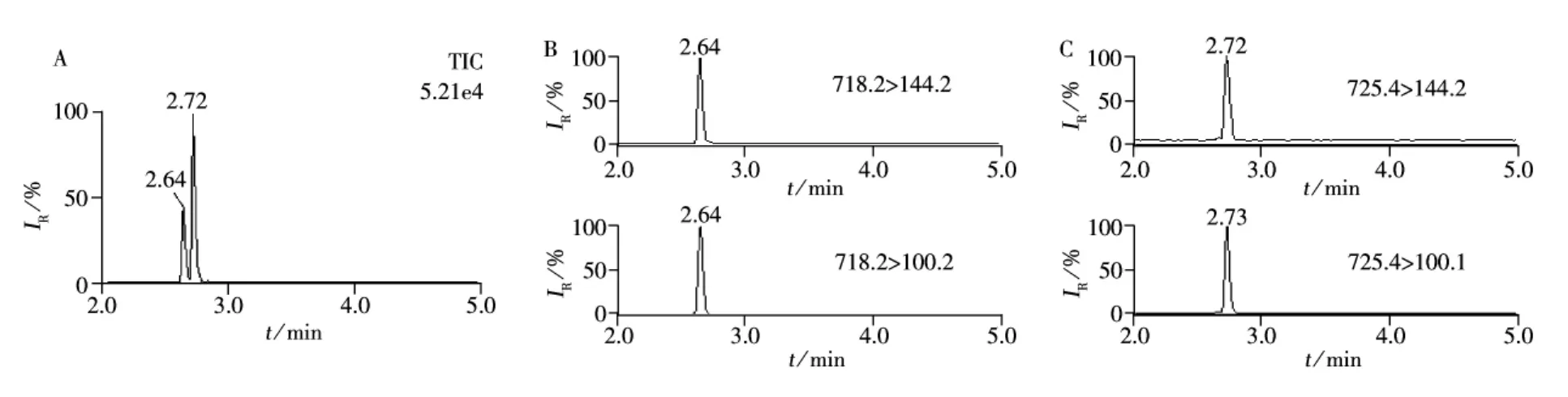
图3 万古霉素和去甲万古霉素混合标准溶液(5.0 μg/L)的总离子流图(A)和提取离子色谱图(B~C)Fig.3 Total ion chromatogram(A)and selective ion chromatograms of norvancomycin(B)and vancomycin(C)mixture standard(5.0 μg/L)
2.3 前处理条件的确定
万古霉素和去甲万古霉素均为两性化合物且结构中含有多个羟基,故易溶于水,微溶于甲醇等有机溶剂。本实验在阴性猪肉样品中添加10 μg/kg的混合标准溶液,对比了以甲醇、乙腈、水、0.1%甲酸水、0.1%甲酸水-乙腈(1∶1)作为提取溶剂的提取效果。结果显示,以纯有机溶剂提取时,提取液澄清,但因溶解度问题,回收率偏低;以纯水相提取时,提取液浑浊,难以离心分离或过滤膜,造成后续净化困难;以0.1%甲酸水-乙腈(1∶1)提取时,提取液较澄清,回收率较高,但提取液中2种化合物的响应值比纯溶剂标准溶液的响应值高,计算得到的回收率均超过100%,说明存在基质效应。因此,本文在进一步比较不同体积比0.1%甲酸水-乙腈混合体系的提取效果时,均与相应的基质匹配标准溶液进行比较,回收率计算结果见图4。可见,0.1%甲酸水的体积分数为50%、60%和70%时,提取效果较好。
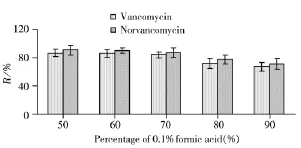
图4 不同配比提取溶剂的提取回收率Fig.4 Comparison of recoveries of extraction solution with different proportions
本文先采用正己烷除去提取液中的脂肪类等非极性杂质。由于动物肌肉组织基质比较复杂,提取液中还含有其它的共萃取杂质干扰检测,并对检测仪器产生一定的污染,因此需进一步净化处理。根据目标物的结构性质,对比了ENVI-Carb(Supelclean,500 mg/6 mL)、Strata - x(Phenomenex,200 mg/3 mL)和LC-C18(CNW,200 mg/3 mL)3种反相固相萃取小柱的净化效果。根据图4,提取效果较好的提取溶剂体系至少含有30%乙腈,此时若直接上样,待测物会直接从反相固相萃取小柱上流出。因此,比较了上样溶液中乙腈含量分别为0%、5%、10%、20%和30%的情况,分别以上述5种配比的0.1%甲酸水-乙腈混合溶液配制10 μg/L待测物混合标准溶液,各取2 mL加入3种预先用甲醇、水活化的固相萃取小柱中,以2 mL对应比例的甲酸水-乙腈溶液淋洗后再以1 mL 50%甲醇淋洗,8 mL甲醇洗脱,收集各个步骤的流出液进行检测,计算回收率(结果见图5)。结果表明上样溶液中乙腈的比例不高于10%时,LC-C18小柱的回收率最好(92%~101%)。

图5 3种固相萃取小柱和不同配比加样溶液的回收率Fig.5 Comparison of recoveries of different SPE and sample solution
综上,本实验选择0.1%甲酸水-乙腈(7∶3)作为提取溶剂,然后取2.0 mL提取液加入4.0 mL 0.1%甲酸水,将乙腈的比例释释至10%,再用LC-C18小柱净化。
2.4 基质效应、线性关系与检出限
采用LC-MS/MS检测复杂基质样品时,常出现基质效应,影响定量分析的准确度和精密度[15]。Matusewski等[16]建立了基质效应的确认方法,即比较阴性基质匹配标准溶液和纯溶剂标准溶液响应值的差异:当比值等于或接近1时,表明不存在基质效应的影响;大于1时,表明存在离子增强作用,反之则表明存在离子抑制作用。
本实验配制浓度范围为0~50 μg/L的系列基质匹配校准工作溶液和纯溶剂标准溶液进行同时测定,分别绘制标准曲线,通过计算基质校准曲线斜率与纯溶剂标准曲线斜率的比值来考察方法的基质效应。结果发现:万古霉素的斜率比值为2.36,去甲万古霉素的斜率比值为1.85,说明存在基质增强效应。在实验中虽尝试采用改善样品前处理[17]以及色谱或质谱条件[18]等手段来减小基质效应,但未获得明显效果,因此采用阴性基质匹配校准溶液进行定量分析。
2种待测物在阴性猪肉基质中的线性方程、线性范围、相关系数见表2,万古霉素和去甲万古霉素在1~50 μg/L范围内线性关系良好,其相关系数(r2)均大于0.99;检出限(LOD,S/N=3)分别为0.5 μg/kg和0.3 μg/kg,定量下限(LOQ,S/N=10)分别为2.0 μg/kg和1.0 μg/kg,表明方法具有较高的灵敏度。

表2 猪肉中2种目标物的线性范围、线性方程、相关系数、检出限和定量下限Table 2 Linear ranges,regression equations,correlation coefficients,LODs and LOQs for 2 analytes in pork
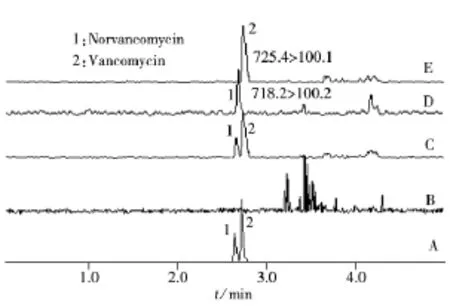
图6 不同样品的TIC色谱图(A~C)及提取离子色谱图(D~E)Fig.6 Total ion chromatograms of different samples(A-C)and selective ion chromatograms(D-E)
2.5 方法的回收率与精密度
选取阴性猪肉样品,按本实验方法进行3个加标水平(LOQ、2LOQ、10LOQ)的回收率和精密度实验,每个加标水平平行测定6次,计算其回收率和相对标准偏差。采用中间浓度的加标水平连续测定5 d,计算日间精密度,实验结果见表3。2种目标物的平均回收率为80%~88%,日内相对标准偏差均不高于13.4%,日间相对标准偏差均不高于12.5%,表明方法的准确度和精密度良好。混合标准溶液的TIC图、阴性猪肉样品的TIC图、添加水平为LOQ的阴性猪肉样品的TIC图及其提取离子色谱图见图6。

表3 猪肉的加标回收率和相对标准偏差Table 3 Recoveries and RSDs of 2 analytes in pork
2.6 实际样品的检测
采用本方法测定了本地市场销售的10例猪肉样品,均未检出2种目标化合物。
3 结论
本实验建立了超高效液相色谱串联三重四极杆质谱(UPLC-MS/MS)测定猪肉中万古霉素和去甲万古霉素的方法,以0.1%甲酸水-乙腈(7∶3)混合体系协同提取,正己烷(乙腈饱和)和LC-C18固相萃取小柱净化。优化了色谱、质谱和前处理条件,有效提高了检测的灵敏度和准确度;方法具有良好的回收率和精密度,适用于猪肉中万古霉素和去甲万古霉素的同时检测。
[1] Ministry of Agriculture of the People s Republic of China Bulletin No.560《Local Standards of Veterinary Drugs Abolished Directory》.(2005 -10 -28).[2005 -11 -01].http://www.moa.gov.cn/zwllm/tzgg/gg/200511/t2005111 7_496523.htm.
[2] Ministry of Health of the People s Republic of China.Food May Be Illegal and Non-food Substances Added to the List of Food Additives Have Been Abused(1-5 Batches Summary).(2011-04-19).[2011-04-22].http://www.moh.gov.cn/publicfiles/business/ht mLfiles/mohwsjdj/s9164/201104/51441.htm.
[3] Deng J J,Wei W.Journal of Military Surgeon in Southwest China(邓晶晶,卫薇.西南军医),2009,11(5):925-926.
[4] Sun X Q,Li Z,Lin W X.Chin.J.Food Hyg.(孙兴权,李哲,林维宣.中国食品卫生杂志),2008,20(3):263-266.
[5] Xiong L,Xin H W,Wu X C,Li Q,Yu A R,Shen Y,Su D.Military Med.J.South China(熊磊,辛华雯,吴笑春,李罄,余爱荣,沈杨,苏丹.华南国防医学杂志),2009,23(6):7,11-13,18.
[6] Zhang H F,Song Q,Dai B,Zhang F C.Pharmaceutical Journal of Chinese People s Liberation Army(张华峰,宋青,戴博,张福成.解放军药学学报),2011,27(1):66-68.
[7] Lin X L,Zhu J P,Fei Y.Chin.J.Clin.Pharm.(林秀丽,朱金平,费燕.中国临床药学杂志),2011,21(4):231-234.
[8] Xu X M.China J.Mod.Med.(徐幸民.中国现代医学杂志),2004,14(14):105-106.
[9] Lin W X,Sun X Q,Tian M,Yu L,Chen X,Li Z.J.Instrum.Anal.(林维宣,孙兴权,田苗,于灵,陈溪,李哲.分析测试学报),2009,28(2):212-215.
[10] Liu J J,Jin F,She Y X,Liu H B,Shi X M,Wang M,Wang J,Xu S Y.Chin.J.Anal.Chem.(刘佳佳,金芬,佘永新,刘洪斌,史晓梅,王淼,王静,徐思远.分析化学),2011,39(5):652-657.
[11] Curren W S S,King J W.J.Chromatogr.A,2002,954(2):41-49.
[12] Inoue K,Mizuno Y,Yoshimi Y.Liquid Chromatogr.,2008,31(9):3871-3878.
[13] Lin W X,Sun X Q,Tian M,Yu L,Xiao S S,Li Z.China Dairy Ind.(林维宣,孙兴权,田苗,于灵,肖珊珊,李哲.中国乳品工业),2009,37(3):46-48.
[14] Su M,Ai L F,Duan W Z,Ha J.Chin.J.Anal.Lab.(苏萌,艾连峰,段文仲,哈婧.分析试验室),2012,31(3):73-77.
[15] Yao M K,Ma B L,Ma Y M.Chin.J.Pharm.Anal.(姚梦侃,马秉亮,马越鸣.药物分析杂志),2010,30(12):2436-2440.
[16] Matuszewski B K,Constanzer M L,Chavez-Eng C M.Anal.Chem.,2003,75(13):3019-3030.
[17] Hernando M D,Suarez-Barcena J M,Bueno M J,Garcia-Reyes J F,Fernandez-Alba A R.J.Chromatogr.A,2007,1155(1):62-73.
[18] Dams R,Huestis M A,Lambert W E,Murphy C M.J.Am.Soc.Mass Spectrom.,2003,14(11):1290-1294.
