2-羟基-3,4,5-三甲氧基苯乙酮的合成
2013-04-06宁志强涂卫军李猛
宁志强,涂卫军,李猛
(1.黑龙江省科学院石油化学研究院,黑龙江哈尔滨 150040;2.空军驻江西地区军事代表室,江西南昌 330024)
2-羟基-3,4,5-三甲氧基苯乙酮的合成
宁志强1,涂卫军2,李猛1
(1.黑龙江省科学院石油化学研究院,黑龙江哈尔滨 150040;2.空军驻江西地区军事代表室,江西南昌 330024)
对半齿泽兰素重要中间体2-羟基-4,5,6-三甲氧基苯乙酮进行合成,以三甲氧基苯为起始原料,经氧化、还原、傅-克酰基化、水解、甲基化5步反应合成了目标产物,并采用单因素实验优化了各步反应条件,总收率48%。采用红外光谱、核磁共振氢谱等检测手段确证了中间体及产物的结构。
2-羟基-4,5,6-三甲氧基苯乙酮;氧化;还原;傅-克酰基化;水解;甲基化
前言
黄酮类化合物半齿泽兰素具有杀螺活性,对预防糖尿病性白内障有利。还有明显的外周镇痛作用,可以抑制多种致痛因子诱发的末梢神经放电等多种功效,除此以外其还具有抗衰老、抗氧化、驱虫等作用[1~3]。但其在天然药物中的含量低,难以满足日益增长的市场需要,2-羟基-4,5,6-三甲氧基苯乙酮是合成半齿泽兰素的关键中间体[4]。
Sastri,A.D.N.[5]等人以2,4,6-三羟基苯乙酮选择性二甲基化得2-羟基-4,6-二甲氧基苯乙酮,Elbs氧化得2,5-二羟基-4,6-二甲氧基苯乙酮,选择性单甲基化得2-羟基-4,5,6-三甲氧基苯乙酮。但其中Elbs氧化收率只有30%,反应总收率仅为14%。段新方[6]等人从3,4,5-三甲氧基苯甲酸出发,经硝化、还原、重氮化、傅克-酰基化、水解制备目标产物。总反应收率为20%,而且我们曾多次实验用3,4,5-三甲氧基苯胺重氮化制备3,4,5-三甲氧基苯酚但重现性不好。Han-WeiChu等人[7],曾采用DMD通过2-羟基-4,6-二甲氧基苯乙酮制备2,5-二羟基-4,6-二甲氧基苯乙酮。但DMD的制备成本较高,而且需要柱层析纯化。此外还曾参考文献[8,9]用3,4,5-三甲氧基苯甲醛氧化制备3,4,5-三甲氧基苯酚,进而制备2-羟基-4,5,6-三甲氧基苯乙酮。但进行多次实验,采用多种氧化条件,氧化3,4,5-三甲氧基苯甲醛都没成功,这主要原因是因为3,4,5-三甲氧基苯甲醛在氧化过程中极易被氧化成醌。
经过对文献的反复筛选,我们以三甲氧基苯为起始原料,经氧化、还原、傅-克酰基化、水解、甲基化5步反应合成关键中间体2-羟基-4,5,6-三甲氧基苯乙酮。

1 实验部分
1.1 主要试剂
1,3,5-三甲氧基苯(工业品,山东默锐化学有限公司),三氟化硼乙醚(分析纯,上海凌峰化学试剂有限公司),硫酸二甲酯(分析纯,太仓市新湖化工助剂厂)。其它试剂均为市售分析纯商品。
1.2 2,6-二甲氧基苯醌的合成
将K3Fe(CN)6(5g,0.015mol)溶解于水25g中,向其中加入1,3,5-三甲氧基苯(42g,0.25mol)和丙酮250g,于室温搅拌,每隔3h,分5次加入过氧化氢(30%,85g,,0.75mol),搅拌15h,析出黄绿色固体,用甲醇脱色,得黄色固体34g,产率81%,纯度99.7%; m.p.250~251℃(lit.[10]240~242℃)。
1.3 2,6-二甲氧基-对羟基苯的合成
将2,6-二甲氧基苯醌(3.1g,0.018mol)、保险粉(12.5g,0.072mol),加入到40mL水中,加热回流30min,溶液呈透明,冷却到10℃左右,冷却30min,过滤,水洗,真空干燥,得白色固体2.8g,产率90.0%,纯度98.0%,m.p.160~161℃(lit.[11]159~161℃)。
1.4 2-羟基-4,6-二甲氧基-5-乙酰基苯乙酮络合物的合成
将(12.6 g,0.074mol)2,6-二甲氧基-对羟基苯悬浮于100 mL二氯甲烷中,加入(56mL,0.59mol)乙酸酐,搅拌下向其中滴入33mL三氟化硼乙醚溶液,加热回流3h,冷却,有固体析出,抽滤,得黄色晶体19.4g,为2-羟基-4,6-二甲氧基-5-乙酰基苯乙酮络合物,产率86.0%,纯度99.0%,m.p.207~210℃。1.5 3,6-二羟基-2,4-二甲氧基苯乙酮的合成
将2-羟基-4,6-二甲氧基-5-乙酰基苯乙酮络合物(14.5g,0.043mol),加入到70mL甲醇中搅拌回流5h,冷却,加入到140mL水中,析出黄色晶体10.1g.,产率100%,纯度96.0%,m.p.156~157℃(lit.[12]162~162.5℃)。
1.6 2-羟基-4,5,6-三甲氧基苯乙酮的合成
3,6-二羟基-2,4-二甲氧基苯乙酮(22.3g,0.105mol),碳酸钾(72.0g,0.522mol),硫酸二甲酯(13.2g,0.105mol),加入到400mL苯中,搅拌加热回流16h,冷却到室温,加入500mL水,搅拌,固体碳酸钾全部溶解,静止分层,水层用冷1∶1盐酸酸化至pH=2,析出黄色固体2.2g,为原料3,6-二羟基-2, 4-二甲氧基苯乙酮。苯层用120mL10%氢氧化钠水溶液萃取,分出水层,用冷1∶1盐酸酸化至pH=2,放入冰箱,冷却固化,得褐色油状固体18.1g,产率76.1%,纯度99.0%,可直接用于下一步反应。m.p. 36~40℃(lit.[60]m.p.33~36℃)。
2 结果与讨论
2.1 2,6-二甲氧基苯醌的合成
2,6-二甲氧基苯醌可以通过1,3,5-三甲氧基苯氧化制取,常用的氧化剂有硝酸或双氧水都可以做氧化剂。我们选用三种氧化体系,进行氧化3,4,5-三甲氧基苯制备2,6-二甲氧基苯醌,分别为HNO3-EtOH、H2O2-AcOH、H2O2-Me2CO-K3Fe(CN)6氧化体系。实验结果见表1。

表1 不同反应体系的对比结果Table1 The comparison of different reaction systems
我们以HNO3-EtOH氧化体系进行氧化,溶液呈黑紫色,得到黑色黏稠液体;从上述实验结果可以看出,采用H2O2-AcOH氧化体系或H2O2-Me2CO-K3Fe(CN)6氧化体系,得到的产率差不多,但从经济成本上考虑,H2O2-AcOH氧化体系所用溶剂冰乙酸无法进行回收而且用量较大,使反应成本增加,而H2O2-Me2CO-K3Fe(CN)6氧化体系所用溶剂丙酮可以进行回收利用,回收率为80%,所以本实验采用H2O2-Me2CO-K3Fe(CN)6氧化体系,氧化1,3,5-三甲氧基苯。
2.2 2,6-二甲氧基-对羟基苯的合成
2,6-二甲氧基-对羟基苯可通过还原2,6-二甲氧基苯醌制取,常用的还原苯醌的方法有用连二亚硫酸钠还原或催化氢化还原。
实验采用连二亚硫酸钠还原2,6-二甲氧基苯醌制备2,6-二甲氧基-对羟基苯,连二亚硫酸钠不稳定,溶于水后逐渐分解为亚硫酸盐和硫代硫酸盐,反应式如下:
2S2O42-+H2O=S2O32-+2HSO3-
连二亚硫酸钠Na2S2O4在稀碱介质中是一种强还原剂,E0=-1.12V。
S2O42-+4OH-=2SO32-+2H2O+2e
用Na2S2O4作还原剂具有还原条件温和,反应快,收率高,产品纯等优点,但是价格较高,保存时易变质。和催化氢化还原反应比较,操作方便。
2.2.1 溶剂的选取
选用连二亚硫酸钠(保险粉)为还原试剂后,对反应溶剂进行选取。实验结果见表2。

表2 各种溶剂对比结果Table 2 The comparison of different solvents
由表可知,选用水为溶剂产率较高,纯度较好,这主要是因为保险粉是在水中分解后起到还原作用,只用水做溶剂,使保险粉分解较完全,还原作用较强,而且用水可以使反应温度升高,使保险粉的还原性增强。
2.2.2 单因素实验
还原试剂和溶剂确定后,对还原反应的还原剂用量、反应时间、反应温度进行单因素实验。结果见表3。

表3 还原反应的单因素效果比较Table3 The comparison of one-factor ex?periments of reduction reaction
由表可知,第一组实验(序号1-3),只改变反应温度,其他条件不变,反应随温度的增加,产率增加,当达到100℃,也就是水的回流温度时产率最高。第二组实验(序号3-5),只改变连二亚硫酸钠的用量,其他条件不变时,当物质的量比为1∶4时,反应效果较好,提高二亚硫酸钠的用量,基本没有变化。第三组实验(序号4、6、7),只改变反应时间,其他条件不变时,反应时间为30min,纯度和收率都很好,延长反应时间,产率虽稍有所提高,但纯度不高,这可能是2,6-二甲氧基-对羟基苯在高温条件下,反应时间过长,又被还原为醌。
综合上述的分析,选用连二亚硫酸钠,溶剂为水,还剂用量为n醌∶n保险粉=1∶4,反应温度为100℃,反应时间为30min。
2.3 3,6-二羟基-2,4-二甲氧基苯乙酮的合成
3,6-二羟基-2,4-二甲氧基苯乙酮可以通过将2,6-二甲氧基-对羟基苯的傅-克酰基化,再水解来合成。在进行傅-克酰基化反应时,选用三氟化硼乙醚-醋酸酐-二氯甲烷的反应条件得到高收率,高纯度的2-羟基-4,6-二甲氧基-5-乙酰基苯乙酮络合物。
文献上相似化合物的水解条件是在盐酸下进行的,我们选用了盐酸或氢氧化钠和适当的溶剂进行水解,结果见表4。

表4 不同水解反应的对比结果Table 4 The comparison of different hydrolysis reaction
由表4可知,直接使用甲醇回流,不用加入酸或碱可以得到高产率、高纯度的3,6-二羟基-2,4-二甲氧基苯乙酮,因为先水解下来的氟硼酸帮助其水解。用盐酸进行水解,原料不能够完全转化,主要是因为先水解下来的硼酸抑制其反应,使原料不能够完全被转化。用氢氧化钠水解,由于氢氧化钠的氧化性,使反应产物被氧化,得到的产品复杂。
2.4 2-羟基-4,5,6-三甲氧基苯乙酮的合成
对3,6-二羟基-2,4-二甲氧基苯乙酮进行甲基化反应,来制备2-羟基-4,5,6-三甲氧基苯乙酮。
2.4.1 甲基化试剂的选择
一般来说,化合物的甲基化采用的甲基化试剂有两种,硫酸二甲酯和碘甲烷,碘甲烷的活性高于前者,首先对甲基化试剂进行选择。以丙酮为溶剂,加热回流。比较硫酸二甲酯和碘甲烷,结果见表5。
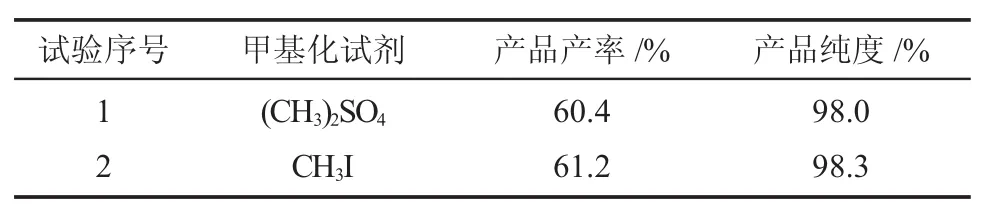
表5 各种甲基化化试剂对比结果Table 5 The comparison of different methylation agents
由表可知,以碘甲烷为甲基化试剂进行反应得到产品的指标要优于硫酸二甲酯,但是差别不是很大,且硫酸二甲酯的价格要低于碘甲烷,所以,选择硫酸二甲酯进行甲基化。
2.4.2 溶剂的选择
甲基化试剂确定之后,我们对溶剂进行选取,比较丙酮与苯的效果,结果见表6。

表6 各种溶剂对比结果Table 6 The comparison of different solvents
由表可知,选用苯为溶剂,增加反应温度,可以使产率提高很多,且纯度较好。
2.4.3 单因素实验
甲基化试剂和溶剂确定后,对甲基化反应的反应时间、催化剂用量、甲基化试剂用量进行单因素试验。结果见表7。

表7 甲基化反应的单因素效果比较Table 7 The comparison of one-factor experiment of methylation reaction
由表可知,第一组实验(序号1-2),只改变硫酸二甲酯的用量其他条件不变时,当物质的量比为1∶1.0时,反应效果较好,提高硫酸二甲酯的用量,产率有所降低,硫酸二甲酯的增加使生成二取代甲基的量增加。第二组实验(序号1、3、4)改变反应时间,当时间为16h,纯度和收率都很好,延长反应时间基本没有大的变化。第三组实验(序号3、5、6)改变催化剂无水碳酸钾的用量,当物质的量比为1:5时,收率和纯度指标都很好,增大比例,没有明显变化。
综合上述的分析,选用硫酸二甲酯甲基化试剂,用量为n原料∶n硫酸二甲酯=1∶1.0,溶剂为苯,催化剂碳酸钾用量为n原料∶n碳酸钾=1∶5,反应时间为16h。
2.5 产物及中间体波谱分析
产物及中间体波谱分析见表8,证实了产物及中间体结构。
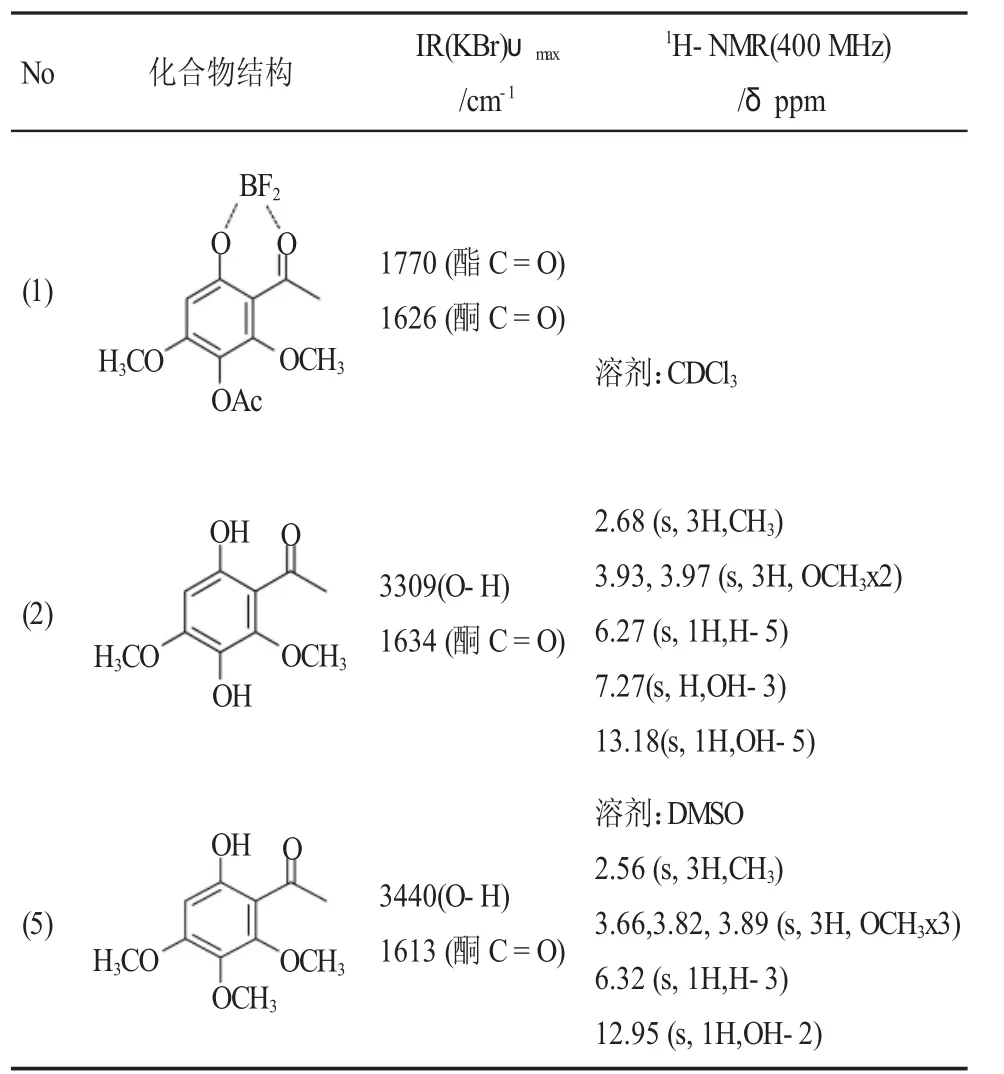
表8 产物及中间体波谱分析Table 8 The spectrum analysis of the intermediate and product
3 结论
以三甲氧基苯为起始原料,经氧化、还原、傅-克酰基化、水解、甲基化5步反应完成其合成,并采用单因素实验优化了各步的反应条件,总收率为48%,高于文献值。
[1]REHMAN A,MALIK A,MEHMOOD S,et al.Phytochemical studies on Indigo fera hetrantha[J].Journal of the Chemical Society of Pakistan,2005,27(4):440~442.
[2]GU L,WENG X.Antioxidant activity and components of Salvia plebeia R.Br.-a Chinese herb[J].Food Chemistry,2001,73(3):299~305.
[3]AYERS SLOAN,ZINK DEBORAH L.Mohn Kenneth.Flavones from Struthiola argentea with anthelmintic activity in vitro[J].Phytochemistry,2007,69(2):541~545.
[4]松浦信.半齿泽兰素的合成方法:JP,50014676A[P].1975-2-15.
[5]SASTRI A D N,SESHADRI T R.Synthesis of 5,6,7-hydroxy flavones and their derivatives[J].Indian Acad.Sci.,1946,23A:242.
[6]段新方,张站斌,段新红.5,3',4'-三羟基-6,7-二甲氧基黄酮的另法全合成[J].有机化学,2003,23(4):353~355.
[7] HAN-WEI CHU, HUAN-TING WU, YEAN-JANG LEE.Regioselective hydroxylation of 2-hydroxychalcones by dimethyldioxirane towards polymethoxylated flavonoids[J].Tetrahedron,2004,60: 2647~2655.
[8]GUZMAN J A,MNDOZA V,GARCIA E,et al.Baeyer-Villiger oxidation of β-Aryl substituted unsaturaied carbonyl compound withhydrogen peroxid and catalytic selenium dioxide[J].Syntheic Communications,1999,25(14):2121~2133.
[9]AMRITAROY,REDDYKR,MONBANTAPK,etal.Hydrogen Peroxide/BoRic Acid:An Efficient Syst for Oxidation of Aromatic Aldhydes and Ketones to Phenols[J].Syntheic Communications,1999, 29(21):3781.
[10]MASAKATSU MATSUMOTO,HISAKO KOBAYASHI.Hexacyano ferrate Catalyzed Oxidation of Trimethoxybenzenes to Dimethoxy-p-benzo-quinones with Hydrogen Peroxide[J].J.Org.Chem.,1985,50:1766~1768.
[11]HIDEAKIOTSUKA,MAMITAKEUCHI,SHOGOINOSHIRIL.Phenolic compounds from coix lachrymal-jobi var.ma-yuen[J].Phytochemistry,1989,28:883~886.
[12]HORTON W J,MASON G S.Boron Trifluoride-Catalyzed Acetylation of Methoxyhydroxybenzenes[J].J.Org.Chem.,1962,27: 830~833.
[13]STOUT MASON G,REICH HANS,HUFFMAN MAX N.4',5,6-Oxygenated flavones and flavanones.[J].J.pharmacy.Sci.1964, 53:192~195.
Synthesis of 2-Hydroxy-4,5,6-trimethoxyacetophenone
NING Zhi-qiang1,TU Wei-jun2and LI Meng1
(1.Institute of Petrochemistry,Heilongjiang Academy of Sciences,Harbin 150040,China;2.The Representative Office of Air Force in Jiang XI,Nan Chang 330024,China)
The intermediate 2-hydroxy-4,5,6-trimethoxyacetophenone was synthesized via 5-step reactions which were oxidation,reduction, Friedel-Crafts acylation,hydrolysis and methylation with using trimethoxybenzene as raw material.And each step was optimized by the single factor test and the yield was 48%.The structure of the intermediates and the desired compounds were confirmed by IR and1H-NMR.
2-Hydroxy-4, 5, 6-trimethoxyacetophenone; oxidation; reduction; Friedel-Crafts acylation; hydrolysis; methylation
TQ 244.2
A
1001-0017(2013)02-0038-04
2012-09-10
宁志强(1971-),男,山东巨野人,高级工程师,主要从事精细化工方面研究。
