β-氨基酸合成研究进展*
2013-03-26曾伟川许瑞安曾庆友1c
曾伟川,许瑞安,曾庆友,1c
(1.华侨大学a.生物医学学院;b.分子药物教育部工程研究中心;c.化工学院,福建泉州 362021)
β-氨基酸近年来越来越受到人们的关注。大量含天然的或非天然的β-氨基酸残基药物已经被广泛应用在医药行业中。首先,广泛使用的抗生素里面含有大量的β-氨基酸残基。世界上最重要的一类抗生素——β-内酰胺环抗生素可以通过 β-氨基酸缩合制备[1];抗生素 Pyloricidin A 和Moiramide A中存在 (S)-β-苯丙氨酸残基;而在蜡样芽胞杆菌中分离出的具有很好抗真菌作用的顺戊霉素也是一种环状的β-氨基酸。其次,一些药物或药物中间体中也含有 β-氨基酸残基[2]。如法国的赛诺菲安万特制药公司治疗急性冠心病药物奥米沙班;德国的默克公司糖尿病新药西他列汀以及美国辉瑞制药公司抗艾滋病新药马拉维罗等。最后,人们研究发现在多肽中引入部分β-氨基酸,其空间二级结构与α-氨基酸组成的多肽非常类似[3]但却有很多独特的性质[4]。
β-氨基酸按侧链所在位置可分为β2-氨基酸,β3-氨基酸和 β2,3-氨基酸(Chart 1)。目前合成 β-氨基酸的方法主要有五种:(1)化学拆分;(2)手性色谱柱拆分;(3)Arndt-Eistert反应;(4)不对称合成(包括不对称加氢,Mannich反应和共轭加成);(5)酶催化。
本文综述了合成β-氨基酸的五种方法,特别是近年来发展迅速的不对称合成和酶催化合成的最新研究进展。

Chart 1
1 化学拆分
化学拆分是一种合成β-氨基酸的经典方法。将消旋的β-氨基酸和手性拆分试剂反应得到一对非对映异构体盐,然后根据两种非对映异构体盐在特定溶液中溶解性差异进行结晶分离。常用的酸性拆分试剂有(+)-酒石酸,(+)-扁桃酸,(+)-樟脑-10-磺酸和 L-(+)-苹果酸等;常用的碱性拆分试剂有(-)-马钱子碱,D-(-)-麻黄碱和(+)-α-苯乙胺等。2008 年,Haycock 等[5]用苯甲醛、丙二酸和碳酸氢铵为原料合成出β-氨基苯丙酸,然后经硫酸催化酯化得到β-氨基苯丙酸甲酯,最后用(L)-酒石酸拆分得(S)-β-氨基苯丙酸甲酯。其中β-氨基苯丙酸甲酯的收率39.6%,化学拆分步骤收率21%,总收率约8.4%。该方法要得到高纯度的产品需要多次结晶。产率低、操作时间长、步骤繁琐等问题限制了其应用。
2 手性色谱柱拆分
手性色谱法具有快速、操作方便和成本低等优点。尽管毛细管电泳法[6]和气相色谱法[7]拆分β-氨基酸也有少量报道,但目前普遍使用的仍是高效液相色谱法。高效液相色谱法又分为间接法和直接法。间接法也叫手性试剂衍生化法,即用一个手性衍生化试剂与一对对映体在柱前反应转化为一对非对映异构体,然后根据非对映异构体物理化学性质不同在普通色谱柱上分离。2004年 Antal等[8]以(S)-N-(4-硝基苯氧基羧基)-苯基丙氨酸甲氧乙酯为手性衍生化试剂分离出18种非天然β-氨基酸。手性固定相法拆分是一种目前应用最广的直接法。其原理是将具有手性识别能力的化合物涂敷或者键合在固定载体上得到手性固定相,通过与被分离物质形成瞬间非对映异构体,利用它们之间的稳定性差异进行分离。2000年,D'Acquarica等[9]分别将大环糖肽类抗生素替考拉宁和A-40,926键合在异氰酸酯修饰的硅胶上分离13种β-氨基酸。分离的氨基酸β-侧链包括脂肪烷烃基、环烷基和芳香基;2002年,péter等[10]用奎宁的氨基甲酸酯衍生物键合在硅胶上成功分离了9 种N-2,4-二硝基苯基或N-3,5-二硝基苯甲基衍生化的 β3-氨基酸;2008年,Berkecz等用冠醚(+)-(18-冠-6)-2,3,11,12-四羧酸先后成功分离了13种β3-氨基酸[11]和10种β-氨基酸[12];2009 年,Ilisz等[13]在环糊精中引入供电子或吸电子基团得到cyclobondⅠ2000 DMP和cyclobond DNP,利用分离物和环糊精的电荷作用,分离各种饱和、不饱和β3-氨基酸和二环β-氨基酸。
3 Arndt-Eistert反应
Arndt-Eistert反应是一种较早使用的用天然α-氨基酸合成β3-氨基酸的方法,包括以下两个步骤:(1)α-氨基酸的氨基经保护后进行羧基活化,之后加入重氮甲烷得重氮甲酮;(2)重氮甲酮经过Wolff重排得到β-氨基酸。因为Wolff重排不改变构型,所以能得到手性β-氨基酸。人们最早用Z-或Boc-保护 α-氨基酸的氨基,用 SOCl2活化为酰氯,然后加入重氮甲烷反应得到重氮甲酮,但是发现外消旋和分解比较严重[14]。如果选用Fmoc做氨基保护基,能很好得到相应的重氮甲酮,收率92%~97%。采用混合酸酐法代替SOCl2活化羧基收率又能得到轻微提高[15]。
Arndt-Eistert同系化只能得到 β3-氨基酸,侧链取代基又受到天然α-氨基酸侧链的限制,加上大量使用有毒的重氮甲烷,严重限制了它的应用。新方法的建立扩大了Arndt-Eistert同系化的应用范围。如Hughes等[16]以 α-氨基酸为底物,先通过 Arndt-Eistert得到 β3-氨基酸,然后再转变为β2,3-氨基酸(Scheme 1)。
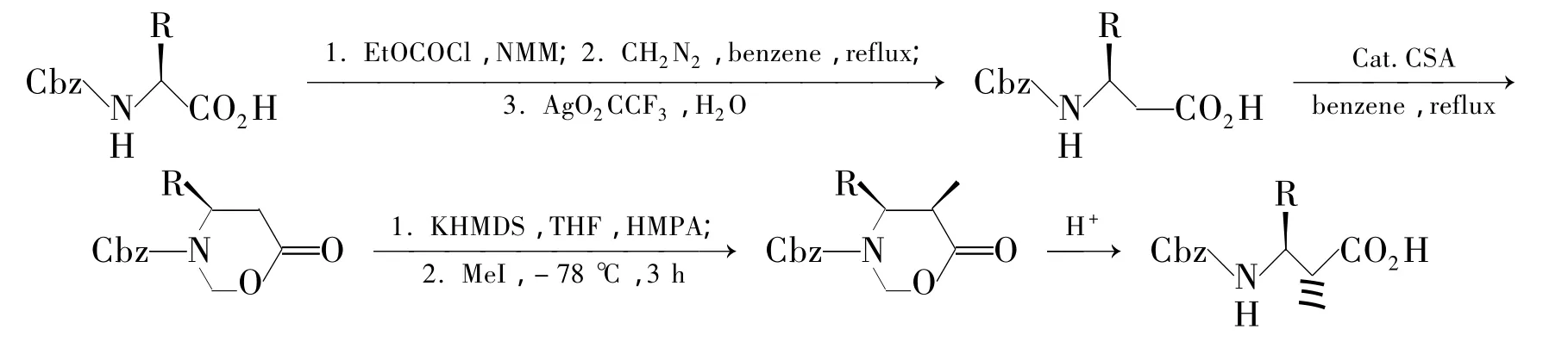
Scheme 1
4 不对称合成
不对称合成是一种非常重要的合成β-氨基酸的方法,研究也较多[17~19]。国内 2002年马治华等[20]也发表过该方面的综述。本文重点介绍2010年后该方法的研究进展。
4.1 催化不对称加氢
催化不对称加氢得到的是 β2-,β3-氨基酸。它原子经济,顺反烯胺都可以作为底物,和Mannich反应相比催化载体用量少(低于1%),且非常容易工业化。1991年,Noyori等[21]首次报道了用 Ru(O2CCH3)2和(R)-1,1'-联萘-2,2'-双二苯膦[(R)-BINAP]催化不对称加氢得到 β-氨基酸,e.e.>90%;随后各种BINAP配体被研究。Hideo等[22]发现BINAP中联芳二面角的度数与对映选择性有很大的关系:键角越小,对映选择性越好;2010年,Zhang等[23]发现引入手性环己烷后配体构象更稳定,在50℃高温下对映选择性达99%。
催化不对称加氢一般都以β-酮酯为原料,主要有以下三种线路:(1)β-酮酯先催化加氢为手性β-羟基酯,然后在羟基上引入易离去基团(如Ts),再用亲核性的氮源取代[24]。(2)β-酮酯先烯胺化,再用乙酰基保护起来后催化加氢,最后脱保护得 β-氨基酸。2011 年,Birch等[25]在铑催化下不对称合成β-氨基酸,能一次工业放大到1.6吨。(3)β-酮酯直接还原氨化。因为直接还原氨化有β-羟基酯副产物,加入钌二醋酸盐能阻止β-羟基酯生成[26]。2011 年,Matsumura等[27]加入钌二醋酸盐,不经保护一步得到 β-氨基酸,收率86%,e.e.95%。医药行业对过渡金属含量有非常严格控制。近年来人们在致力寻找有机催化剂代替过渡金属。2012年,Simon等[28]用咪唑衍生物催化得到 β-氨基酸,收率 76%,e.e.91%。但是有机催化剂催化能力和选择性效果有待进一步增强。
4.2 Mannich 反应
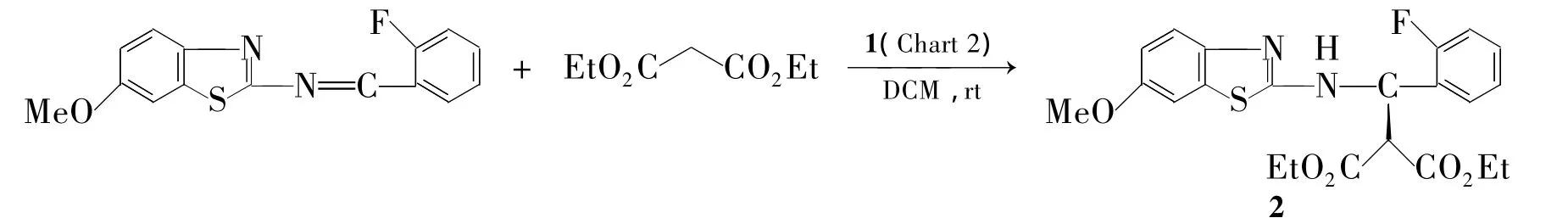
Mannich 反应因能得到各种 β2-,β3-,β2,3-氨基酸而被非常广泛的使用。催化剂可分为金属催化剂和有机催化剂。金属催化剂研究较早,包括金属部分和配体部分,配体提供一个手性环境。使用的金属有 Zn[29],Fe[30],In[31],La[32],Ni[33],Zr[34],Cu[35,36]等。由于重金属有毒,不适合于医药行业。近年来人们更多地开发有机催化剂,常见的 有 DBU[37],金 鸡 纳 碱[38],硫 脲[39],联 萘酚[40],磷酸[41,42]和脯氨酸[43,44]等。其机理是通过与亲电体形成氢键去激活底物,促使过渡态形成。
2012年,Weihua等[45]使用双官能团催化剂1(Chart 2,一端连金鸡纳碱,一端连硫脲),金鸡纳碱活化丙二酸二乙酯,硫脲活化亚胺,起到双重催化作用。能催化一些活性不高(丙二酸二乙酯等)的反应,扩大了Mannich反应使用范围。带苯并噻唑的亚胺与丙二酸二乙酯在1催化下合成高光学纯度的β-氨基酸2,收率91%,纯度98.5%。对催化剂优化发现,在苯环上引入吸电子基团能提高产率和光学纯度(Scheme 2)。由于亚胺(如N-Boc-亚胺)对水和空气都相当敏感,即使在低温短时间内也迅速自发异构化[46]。2010年,Luca等[47]以 α-氨基砜为原料,在 L-脯氨酸和 KF 碱催化下,原位先得到亚胺,然后立即进行Mannich反应,再氧化,不经分离一锅法制得β-氨基酸(3),产率 92%,e.e.>99%(Scheme 3)。
Mannich反应还能引入一些难引入的功能基团。2010年,Haile等[48]以廉价的甘氨酸酯为原料,在金鸡纳碱硫脲双官能团催化剂4(Chart 3)催化下合成得到各种 α,β-二氨基酸,产率62% ~98%,e.e.99%。2012 年,Kano 等人[49]以Z-或Boc-氨基乙醛为原料制备α,β-二氨基酸甲酯。氟原子因其独特的性质被广泛用于药物中[50]。2010年,Yuanhang等人[51]用胍 5(Chart 3)催化不对称合成含氟的 β-氨基酸 6,产率 99%,e.e.>99%(Scheme 4)。
4.3 共轭加成
(1)亲核性碳原子共轭加成
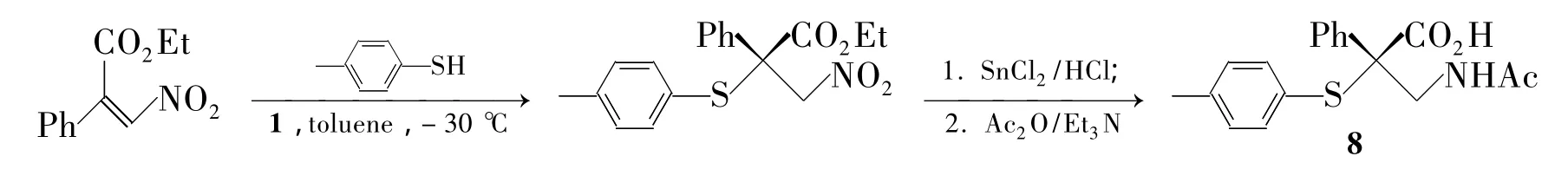
亲核性碳原子共轭加成得到 β2-或 β2,2-氨基酸。用碳负离子加成到硝基烯,烯醛或二酯烯等受体上,再通过还原或氧化得到 β-氨基酸[52~56]。2010年,Pablo等[57]将有机相转移催化剂和金鸡纳碱结合得到双官能团催化剂4(Chart 3),双重功效催化TMSCN加成到硝基烯上,高效得到7(Scheme 5)。Wang 等[58]将受体用 α,β-不饱和二酯替代硝基烯,亲核试剂CNCOOEt替代TMSCN。受体价格更低廉,亲核试剂毒性更低;2012年,Lin等[59]用 Ti(OiPr)4催化加成氰基到硝基烯上,通过锌粉还原,酸水解等步骤得到β-氨基酸,产率90%。β2,2-氨基酸因底物位阻大和反应可逆导致合成困难。Haihua等[60]用双官能团催化剂1成功把对苯硫酚加成到底物上得到β2,2-氨基酸8,收率 96%,e.e.94%(Scheme 6)。
Scheme 2

Scheme 3

Scheme 4

Scheme 5
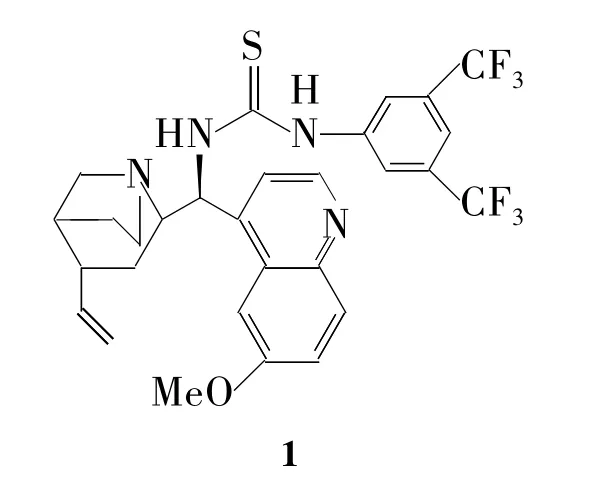
Chart 2

Chart 3
(2)亲核性氮原子共轭加成
亲核性氮原子共轭加成得到β3-氨基酸。用亲核性胺(芳香胺、羟胺、氨基锂等)加成到α,β-不饱和羰基化合物上。该法对受体活性有较高要求。金属-配体催化不能以活性较低的α,β-不饱和酯为受体;原料用手性氨基锂或有机相转移催化剂催化可以加成到α,β-不饱和酯上。金属-手性配体催化常用的配体是BINAP。镁、铜、锂、钐等金属[61~64]都用作催化剂。2011 年,Dorian等[65]用碘化钐不对称加成苯甲羟胺到 α,β-不饱和羰基化合物,氨源由以前的芳香胺扩大到羟胺。
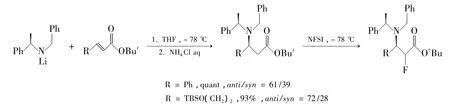
手性氨基锂被广泛应用到合成β-氨基酸中。手性氨基锂加成到各种芳香或脂肪α,β-不饱和酯上,再脱氨保护基和酯基即可得到手性β-氨基酸。2005年,Stephen等[66]对这方面研究进展有详细的总结;最近,Peter等[67]用此路线在 α-位引入 F 原子(Scheme 7);2012 年,Weiβ 等[68]成功使用有机手性相转移催化剂9(Chart 4)不对称加成亲核性氮到α,β-不饱和酯上,收率86%。遗憾的是对映选择性较低(37%),还需要进一步改进(Scheme 8)。
5 酶催化
生物催化α-氨基酸的酶很多,但仅有极少部分酶才能催化 β-氨基酸。2006年,Arto等[69]综述过生物催化β-氨基酸,本文重点介绍生物催化2006年后新的研究进展。
Scheme 6
Scheme 7

Scheme 8

Scheme 9
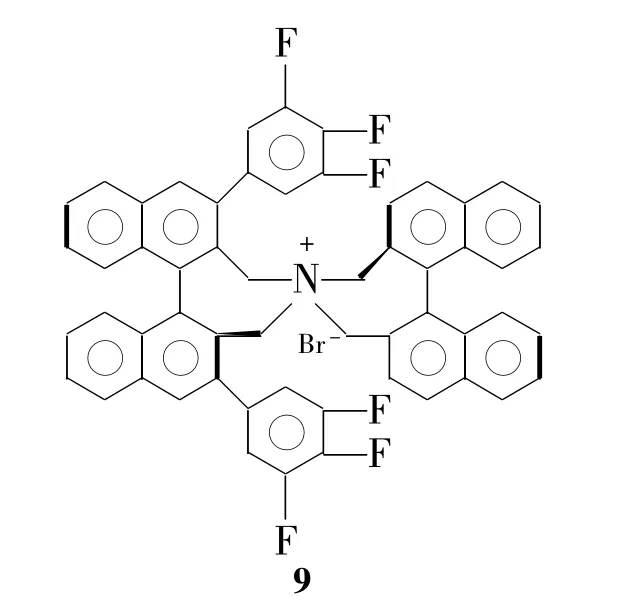
Chart 4
5.1 酰胺酶
(1)青霉素G酰化酶
青霉素G酰化酶(PGA)是最早研究的一种能生物催化β-氨基酸的酶。1995年,Soloshonok等[70]用 PGA 在 PH 7.0 ~8.5 之间制备了一系列β-氨基酸。该酶现已广泛应用于工业生产,并固定在各种载体上重复回收利用,以降低工业成本[71]。PGA 能够很好的得到 β3-,β2,3-氨基酸,转化率近 50%,e.e.95%。但是转化 β2-氨基酸效果差,且氨基保护基只能是苯乙酰基。
(2)转肽酶
转肽酶能识别特异性酰胺键或多肽,催化不同的转肽反应。2009年,Tobias等[72]用 3-2W4 BapA,Y2 BapA,DmpA三种转肽酶在 pH 8,37℃下催化得到β3-氨基酸。酶和取代基种类不同,收率也有所不同,最高收率达48%,e.e.98%。
(3)乙内酰脲酶
乙内酰脲酶能水解二氢尿嘧啶。2011年,Maeve等[73]从 Vigna Angularis中得到的乙内酰脲酶催化各种5-取代尿嘧啶,再用亚硝酸脱保护得到β-氨基酸。研究发现5-位取代基为芳香基效果最好(Scheme 9)。
5.2 氨基转移酶
氨基转移酶(TAs)具有催化底物的成本很低,不需要外部辅助因子,有高的对映选择性和反应速率等优点。有消旋拆分和不对称转化两种方法(Scheme 10)。

Scheme 10
Scheme 11
(1)以消旋β-氨基酸为底物进行消旋拆分。2011年,Han等[74]用消旋芳香氨基酸与丙酮酸为原料,在 TAs催化下得到 L-丙氨酸和(R)-β-芳香氨基酸,收率约 50%,e.e.>99%。
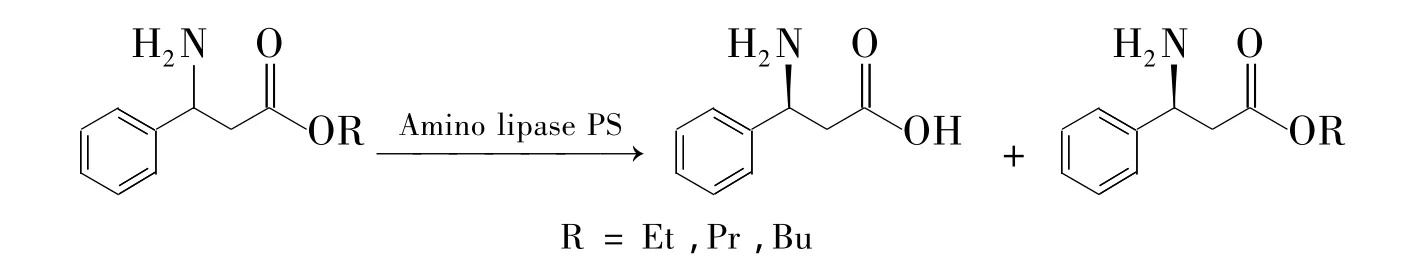
(2)以β-酮酸为底物进行酶不对称转化(理论产率为100%)。β-酮酸非常容易通过形成环过渡态而脱去羧基[75],用稳定的β-酮酯为原料制备β-酮酸,然后立即酶催化得到β-氨基酸是一种好方法。2007年,Juhan等[76]用脂肪酶把 β-酮酯转化为β-酮酸,再不对称转化得到β-氨基酸。遗憾的是收率只有20%,e.e.99%。天然得到的氨基转移酶TAs对短链脂肪β-氨基酸(β-丙氨酸,β-氨基丁酸等)有很高的催化效率。但是对芳香β-氨基酸(β-苯丙氨酸)没有活性[77]。而且,酮基的邻位取代基体积不能大于甲基,否则催化无法进行[78]。为了扩大底物范围,很多高效筛选新型TAs的方法已经建立起来并且取得了一些成功[79~82]。如 Christopher等[83]筛选出新的 TAs转移邻位带苯环的酮基为氨基得到(S)-西他列汀,收率92%,对映选择性高达99.95%。
5.3 脂肪酶
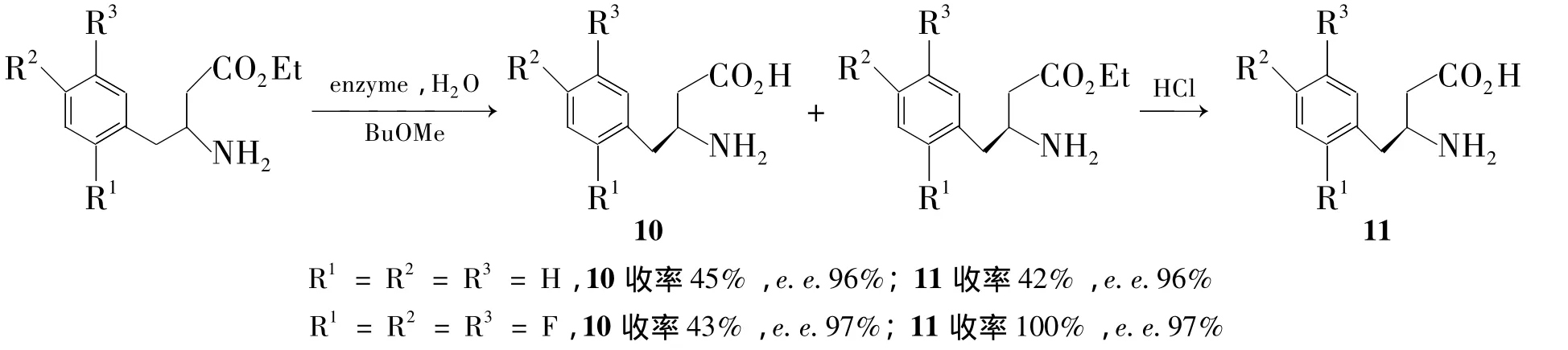
2010年,Gabor等[84]把 Lipase PS 固定化到硅藻土上拆分 β-苯基-β-氨基酸乙酯,得到(R)-β-苯基-β-氨基酸(10),收率45%,e.e.96%。未被酶水解的脂经盐酸水解后也能得到(S)-β-苯基-β-氨基酸(11),收率42%,e.e.96%(Scheme 11)。
2011 年,Grayson 等[85]将消旋 β-苯基丙氨酸丙酯溶于甲基叔丁基醚中,Lipase PS水溶液加入形成油水两相,在pH 8时Lipase PS水解消旋β-苯基丙氨酸丙酯产生(S)-β-苯基丙氨酸。利用(S)-β-苯基丙氨酸不溶于油水两相沉淀出来,收率42%,e.e.99.5%,纯度高。该方法适合工业化生产,文献报道一次生产量178 kg(Scheme 12)。
动态动力学拆分[86]是指在催化剂催化下底物原位消旋,酶消耗掉的对映体得到源源不断补充而得到更多的产物,理论产率达100%。2010年,Mozaffar等[87]用钌催化剂12(Chart 5)消旋 3-氨基-3-苯基丙酸酯,不断的补充酶拆分掉的左旋体而实现动态动力学拆分。作者还将南极假丝酵母脂肪酶(CALA)固定在硅介孔泡沫材料(MCF)上增加酶热稳定性,能耐90℃高温,克服了大多数酶不耐高温的缺点,收率 85%,e.e.89%(Scheme 13)。
5.4 变位酶
苯丙氨酸氨基变位酶(PAM)能催化β-苯丙氨酸为α-苯丙氨酸。将消旋体中的(R)-β-苯丙氨酸转化为(S)-α-苯丙氨酸,从而获得未转化的(S)-β-苯丙氨酸。但PAM催化是个平衡反应,转化不完全。苯丙氨酸解氨酶(PAL)能转化α-苯丙氨酸到肉桂酸。2010年,Bian等[88]利用PAL酶转化掉α-苯丙氨酸打破平衡,得到高纯度的(S)-β-苯丙氨酸,收率 48%,e.e.99%(Scheme 14)。
研究还发现PAM在转化β-苯丙氨酸到α-苯丙氨酸过程中检测到(E)-肉桂酸的存在。根据这启示,2009年,Miktor等[89]用肉桂酸为底物PAM催化得到约1∶1的α-氨基酸和β-氨基酸混合物,e.e.均在99%以上。在苯环各个位置引入不同取代基,两种混合物的比例不一样。在邻位引入取代基主要得到α-氨基酸,在对位引入供电子基主要得到β-氨基酸;为了进一步提高β-氨基酸含量,2012年,Bian等[90]筛选几种 PAM 的突变型酶进行催化并探讨其机制。研究发现酶Q319M效果不错,最高收率达99%,e.e.99%。
Scheme 12
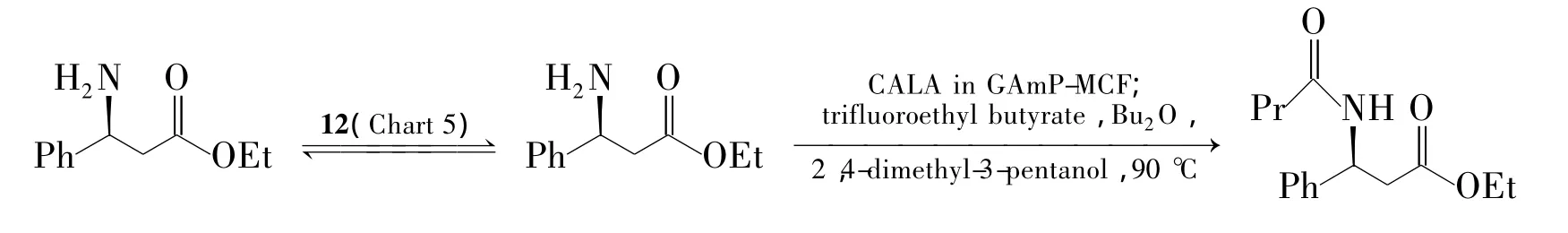
Scheme 13
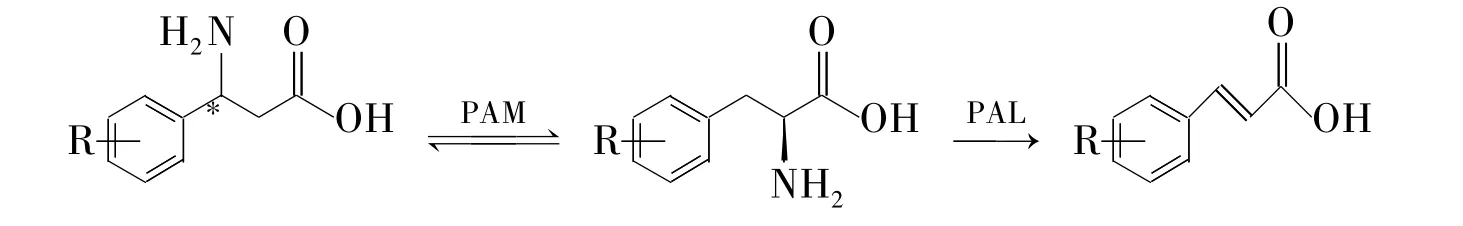
Scheme 14

Chart 5
6 结语
化学拆分和手性色谱柱拆分是较早使用的拆分方法。化学拆分需要手性拆分剂,且操作繁琐;手性色谱柱拆分在衍生化过程中容易引入副产物,且手性柱价格昂贵。β3-氨基酸且是天然侧链的一般用 Arndt-Eistert反应制备,因为天然 α-氨基酸方便易得且成本低廉,加之该反应能规模化生产。如果侧链为非天然的β-氨基酸,一般用催化加氢。它原子经济,易于工业化,顺反烯胺都可以作为原料。但过渡金属的使用,毒且贵,不能用于制药行业。Mannich反应能原子经济的得到各种类型的β-氨基酸。近年来已开发出一些较好的有机催化剂,在追求无毒、对水和空气稳定、一锅法制备等方面都取得长足发展。共轭加成对亲核试剂或受体活性有较高要求,限制了它的广泛应用,目前正致力于扩大其底物范围。
消旋体拆分和化学合成得到的产品总体来讲光学纯度都偏低,达不到制药行业中一些高要求标准。生物酶制备的β-氨基酸有非常高的光学纯度,有的高达99.95%。且反应条件温和,不需要配体,正越来越受到科学家的广泛关注。动态动力学拆分克服了动力学拆分理论产率只有50%的缺陷,酶不对称转化更是展现出非常光明的前景。但是,酶只能对特定的底物进行催化,限制了其广泛的应用。如何寻求一种能广泛催化各种β-氨基酸的酶是今后酶研究的重点。近年来,一些高效率筛选酶的方法已经建立起来并取得一些可喜进展。
[1]Magriotis P A.Recent progress in the enantioselective synthesis of β-lactams:Development of the first catalytic approaches[J].Angew Chem Int Ed,2001,40(23):4377-4379.
[2]Grayson J L,Roos J,Osswald S.Development of a commercial process for(S)-β-phenylalanine[J].Org Process Res Dev,2011,15(5):1201 -1206.
[3]Sadowsky J D,Murray J K,Tomita Y.Exploration of backbone space in foldamers containing α- and β-amino acid residues:Developing protease-resistant oligomers that bind tightly to the BH3-recognition cleft of Bcl-XL[J].ChemBioChem,2007,8(8):903 -916.
[4]Seebach D,Gardiner J.β-peptidic peptidomimetics[J].Accounis of chemical research,2008,41(10):1366-1375.
[5]Haycock-Lewandowski S J,Wilder A,Ahman J.Development of a bulk enabling route to maraviroc(UK-427,857),a CCR-5 receptor antagonist[J].Org Process Res Dev,2008,12(6):1094 -1103.
[6]Kuhn R,Erni F,Bereuter T,et al.Chiral recognition and enantiomeric resolution based on host-guest complexation with crown ethers in capillary zone electrophoresis[J].Anal chem,992,64(22):2815 -2820.
[7]Heid C B,Robbins D K.Investigation of liquid and gas chromatography techniques for separation of diastereomers of β-(α-methylbenzyl)amino isobytyric acid[J].J Chromatogr Sci,2003,41:422 -427.
[8]Péter A,Árki A,Vékes E,et al.Direct and indirect high-performance liquid chromatographic enantioseparation pf β-amino acids[J].ChromatogrA,2004,1031:171-178.
[9]D'Acquarica I,Gasparrini F,Misiti D,et al.Application of a new chiral stationary phase containing the glycopeptide antibiotic A-40,926 in the direct chromatographic resolution of β-amino acids[J].Tetrahedron:Asymmetry,2000,11:2375 -2385.
[10]Péter A.Direct high-performance liquid chromatographic enantioseparation of apolar β-amino acids on a quinine-derived chiral anion-exchanger stationary phase[J].Chromatography A,2002,955:141 - 150.
[11]Berkecz R,Ilisz I,Fulup F J,et al.High-performance liquid chromatographic enantioseparation of β-3-homo-amino acid stereoisomers on a(+)-(18-crown-6)-2,3,11,12-tetracarboxylic acid - based chiral stationary phase[J].Chromatogr A,2008,1189:285-291.
[12]Berkecz R,Ilisz I,Patai Z.LC enantioseparation of β-amino acids on a crown ether - based stationary phase[J].Chromatographia,2008,68:S13 - S18.
[13]Ilisz I,Berkecz R,ForrÕE.The role of π-acidic and π-basic chiral stationary phases in the high-performance liquid chromatographic enantioseparation of unusual β-aminc acids[J].Chirality,2009,21:339 -348.
[14]Hodson D,Holt G,Wall D K.Diazoketones from the interaction of diazoalkanes with carboxylic acid-dicyclohexylcarbodi-imide mixtures[J].Journal of the Chemical Society C:Organic,1970,7:971 -973.
[15]Guichard G,Abele S,Seebach D.Preparation ofNFmoc-protected β2-and β3-amino acids and their use as building blocks for the solid-phase synthesis of βpeptides[J].Helv Chim Acta,1998,81(2):187 -206.
[16]Sleebs B E,Hughes A B.Diastereoselective synthesis of α-methyl and α-hydroxy-β-amino acids via 4-substituted-1,3-oxazinan-6-ones[J].J Org Chem,2007,72(9):3340 -3352.
[17]Liu M,Sibi M P.Recent advances in the stereoselective synthesis of β-amino acids[J].Tetrahedron,2002,58:7991 -8035.
[18]Sleebs B E,Nguyen T T,Hughes A B,et al.Recent advances in stereoselective synthesis and application of β-amino acids[J].Organic preparations and procedures international,2009,41:429 -478.
[19]Weiner B,Szymański W,Janssen D B,et al.Recent advances in the catalytic asymmetric synthesis of β-amino acids[J].Chem Soc Rev,2010,39:1656 -1691.
[20]马治华,赵永华,王建波.β-氨基酸不对称合成研究得新进展[J].有机化学,2002,22(11):807 -816.
[21]Lubell W D,Kitamura M,Noyori R.Enantioselective synthesis of β-amino acids based on BINAP-ruthenium(Ⅱ)catalyzed hydrogenation[J].Tetrahedron:Asymmetry,1991,2(7):543 -554.
[22]Shimizu H,Nagasaki I,Matsumura K,et al.Developments in asymmetric hydrogenation from an industrial perspective[J].Acc Chem Res,2007,40:1385 -1393.
[23]Zhang X W,Huang K X,Hou G H,et al.Electrondonating and rigid P-stereogenic bisphospholane ligands for highly enantioselective Rhodium-catalyzed asymmetric hydrogenations[J].Angew Chem Int Ed,2010,49:6421 -6424.
[24]Hansen K B,Balsells J,Dreher S,et al.First generation process for the preparation of the DPP-Ⅳ inhibitor Sitagliptin[J].J Org process Res Dev,2005,9(5):634-639.
[25]Birch M,Challenger S,Crochard J P,et al.The development of a practical multikilogram synthesis of the chiral β-amino acid imagabalin hydrochloride(PD-0332334)via asymmetric hydrogenation[J].Org process Res Dev,2011,15:1172 -1177.
[26]Noyori R,Ohkuma T,Kitamura M,et al.Asymmetric hydrogenation of β-keto carboxylic esters.A practical,purely chemical access to β-hydroxy esters in high enantiomeric purity[J].J Am Chem Soc,1987,109(19):5856-5858.
[27]Matsumura K,Zhang X Y,Hori K,et al.Practical,catalytic enantioselective hydrogenation to synthesizeN-unprotected β-amino esters[J].Org Process Res Dev,2011,15:1130 -1137.
[28]Jones S,Li X F.Synehesis of chiral β-amino acid derivatives by asymmetric hydrosilylation with an imidazole derived organocatalyst[J].Tetrahedron,2012,68:5522 -5532.
[29]Trost B M,Terrell L R.A direct catalytic asymmetric Mannich-type reaction to syn-amino alcohols[J].J Am Chem Soc,2003,125(2):338 -339.
[30]Yamashita Y,Ueno M,Kuriyama Y,et al.Catalytic asymmetric Mannich-type reactions using a novel chiral iron complex[J].Adv Synth Catal,2002,344(9):929-931.
[31]Harada S,Handa S,Matsunaga S,et al.Direct catalytic asymmetric Mannich-Type reactions ofN-(2-hydroxyacetyl)pyrrole as an ester-equivalent donor[J].Angew Chem Int Ed,2005,44(28):4365 -4368.
[32]Morimoto H,Aoyama N,Matsunaga S,et al.Lanthanum aryloxide/pybox-catalyzed direct asymmetric Mannich-type reactions using a trichloromethyl ketone as a propionate equivalent donor[J].J Am Chem Soc,2007,129(31):9588 -9589.
[33]Chen Z,Morimoto H,Matsunaga S,et al.A benchstable homodinuclear Ni2-schiff base complex for catalytic asymmetric synthesis of α-tetrasubstituted antiα,β-diamino acid surrogates[J].J Am Chem Soc,2008,130(7):2170 -2171.
[34]Ihori Y,Yamashita Y,Kobayashi S.Chiral zirconium catalysts using multidentate BINOL derivatives for catalytic enantioselective Mannich-type reactions;ligand optimization and approaches to elucidation of the catalyst structure[J].J Am Chem Soc,2005,127(44):15528-15535.
[35]Salter M M,Kobayashi J,Shimizu Y,et al.Directtype catalyticthree-componentMannich reactions leading to an efficient synthesis of α,β-diamino acid derivatives[J].Org Lett,2006,8(16):3533 - 3536.
[36]Suzuki Y,Yazaki R,Shibasaki M.Direct catalytic asymmetric Mannich-type reaction of thioamides[J].Angew Chem Int Ed,2009,48(27):5026 -5029.
[37]Utsumi N,Kitagaki S,Barbas C F.Organocatalytic Mannich-type reactions of trifluoroethylthioesters[J].Org Lett,2008,10(16):3405 -3408.
[38]Ricci A,Pettersen D,Bernardi L,et al.Organocatalytic enantioselective decarboxylative addition of malonic half thioesters to imines[J].Adv Synth Catal,2007,349(7):1037 -1040.
[39]Wenzel A G,Jacobsen E N.Asymmetric catalytic Mannich reactions catalyzed by urea derivatives:Enantioselective synthesis of β-aryl-β-amino acids[J].J Am Chem Soc,2002,124(44):12964 -12965.
[40]Hasegawa A,Naganawa Y,Fushimi M.Design of bronsted acid-assisted chiral bronsted acid catalyst bearing a bis(triflyl)methyl group for a Mannichtype reaction[J].Org Lett,2006,8(15):3175 -3178.
[41]Itoh J,Fuchibe K,Akiyama T.Preparation of β-amino esters by a chiral Bronsted acid-catalyzed Mannich-type reaction[J].Synthesis,2008,8:1319 -1322.
[42]Yamanaka M,Itoh J,Fuchibe K,et al.Chiral Bronsted acid catalyzed enantioselective Mannich-type reaction[J].J Am Chem Soc,2007,129(21):6756 -6764.
[43]Yang J W,Stadler M,ListB.Proline-catalyzed Mannich reaction of aldehydes withN-Boc-imines[J].Angew Chem Int Ed,2007,46(4):609 -611.
[44]Chandler C,Galzerano P,List B.The proline-catalyzed double Mannich reaction of acetaldehyde withN-Boc imines[J].Angew Chem Int Ed,2009,48(11):1978-1980.
[45]Li W H,Song B,Bhadury P S,et al.Chiral cinchona alkaloid-derived thiourea catalyst for enantioselective synthesis of novel β-amino esters by Mannich reaction[J].Chirality,2012,24:223 -231.
[46]Pinaki S B,Song Y,Bao A S.Catalytic synthesis of optically active β-amino acid derivatives[J].Current Organic Synthesis,2012,9:695 -726.
[47]Deiana L,Zhao G L,Dziedzic P,et al.One-pot highly enantioselective catalytic mannich-type reactions between aldehydes and stable α-amido sulfones:Asymmetric synthesis of β-amino aldehydes and β-amino acids[J].Tetrahedron letters,2010,51:234-237.
[48]Zhang H,Syed S,BarbasⅢ C F.Highly enantioand diastereoselective mannich reactions of glycine schiff bases with in situ generatedN-Boc-imines catalyzed by a cinchona alkaloid thiourea[J].Org Lett,2010,12(4):708 -711.
[49]Kano T,Sakamoto R,Akakura M,et al.Stereocontrolled synthesis of vicinal diamines by organocatalytic asymmetric mannich reaction ofN-protected aminoacetaldehydes:Formal synthesis of(- )-agelastatin A[J].J Am Chem Soc,2012,134(17):7516 - 7520.
[50]Poisson T,Belhomme M C,Pannecoucke X.Indiumpromoted Reformatsky reaction:A straightforward access to β-Amino and β-hydroxy α,α-difluoro carbonyl compounds[J].J Org Chem,2012,77(20):9277 -9285.
[51]Pan Y H,Zhao Y J,Ma T,et al.Enantioselective synthesis of α-fluorinated β-amino acid derivatives by an asymmetric Minnich reaction and selective deacylation/decarboxylation reactions[J].Chem Eur J,2010,16:779 -782.
[52]Eilitz U,Leβmann F,Seidelmann O,et al.Stereoselective synthesis of β2-amino acids by Michael addition of diorgano zinc reagents to nitro acrylates[J].Tetrahedron:Asymmetry,2003,14(2):189 -191.
[53]Eilitz U,Leβmann F,Seidelmann O,et al.Stereoselective Michael addition of trimethyl aluminium to nitro acrylates:A route to 2-methyl-3-amino propionic acid[J].Tetrahedron:Asymmetry,2003,14(20):3095-3097.
[54]Sammis G M,Jacobsen E N.Highly enantioselective,catalytic conjugate addition of cyanide to α,βunsaturated imides[J].J Am Chem Soc,2003,125(15):4442-4443.
[55]Duursma A,Minnaard A J,Feringa B L.Highly enantioselective conjugate addition of dialkylzinc reagents to acyclic nitroalkenes:A catalytic route to β2-amino acids,aldehydes,and alcohols[J].J Am Chem Soc,2003,125(13):3700 -3701.
[56]Rimkus A,Sewald N.First synthesis of a β2-homoamino acid by enantioselective catalysis[J].Org Lett,2003,5(1):79 -80.
[57]Bernal P,Fernndez R,Lassaletta J.Organocatalytic asymmetric cyanosilylation of nitroalkenes[J].Chemistry-A Europern Journal,2010,16(26):7714 -7718.
[58]Wang J,Li W,Liu Y,et al.Asymmetric cyanation of activated olefins with ethyl cyanoformate catalyzed by a modular titanium catalyst[J].Org Lett,2010,12(6):1280-1283.
[59]Lin L,Yin W,Fu X,et al.Asymmetric cyanation of nitroalkenes catalyzed by a salen-titanium catalyst[J].Org Biomol Chem,2012,10,83 -89.
[60]Haihua L,Fugen Z,Xianggao M.Enantioselective Michael reactions of β,β-disubstituted nitroalkenes:A new approach to β2,2-amino acids with hetero-quaternary stereocenters[J].Org Lett,2009,11(17):3946-3949.
[61]Sibi M P,Prabagaran N,Ghorpade S G,et al.Enantioselective synthesis of α,β-disubstituted-β-amino acids[J].J Am Chem,Soc,2003,125(39):11796 -11797.
[62]Reboule I,Gil R,Collin J.Enantioselective conjugate addition of aromatic amines toN-alkenoyloxazolidinones catalyzed by Iodido(binaphtholato)samarium[J].Eur J Org Chem,2008,2008(3):532 -539.
[63]Palomo C,Oiarbide M,Halder R,et al.Catalytic enantioselectiveconjugate addition ofcarbamates[J].J Am Chem Soc,2004,126(30):9188 -9189.
[64]Yamagiwa N,Qin H,Matsunaga S,et al.Lewis acid-Lewis acid heterobimetallic cooperative catalysis:mechanistic studies and application in enantioselective Aza-Michael reaction[J].J Am Chem Soc,2005,127(38):13419 -13427.
[65]Didier D,Meddour A,Collin J.Samarium iodobinaphtholate:An efficient catalyst for enantioselective Aza-Michael additions ofO-benzylhydroxylamine toN-alkenoyloxazolidinones[J].Eur J Org Chem,2011,2011(14):2678 -2684.
[66]Davies S G,Smith A D,Price P D.The conjugate addition of enantiomerically pure lithium amides as homochiral ammonia equivalents:Scope,limitations and synthetic applications[J].Tetrahedron:Asymmetry,2005,16(17):2833 -2891.
[67]Duggan P J,Johnston M,March T L.Enantioselective synthesis of α-fluoro-β3-amino esters:Synthesis of enantiopure,orthogonally protected α-fluoro-β3-lysine[J].J Org Chem,2010,75(21):7365 -7372.
[68]Weiβ M,Borchert S,Rémond E,et al.Asymmetric addition of a nitrogen nucleophile to an enoate in the presence of a chiral phaes-transfer catalyst:A novel approach toward enantiomerically enriched protected β-amino acids[J].Heteroatom Chemistry,2012,23(2):202-209.
[69]Liljeblad A,Kanerva L T.Biocatalysis as a profound tool in the preparation of highly enantiopure β-amino acids[J].Tetrahedron,2006,62(25):5831 -5854.
[70]Soloshonok V A,Fokina N A,Rybakova A V,et al.Biocatalytic approach to enantiomerically pure β-amino acids[J].Tetrahedron:Asymmetry,1995,6(7):1601-1610.
[71]Kallenberg A I,Rantwijk F V,Sheldon R.Immobilization of penicillin G acylase:The key to optimum performance[J].Adv Synth Catal,2005,347(7 - 8):905-926.
[72]Heck T,Seebach D,Osswald S,et al.Kinetic resolution of aliphatic β-amino acid amides by β-amin-opeptidases[J].ChemBioChem,2009,10(9):1558-1561.
[73]Neill M,Hauer B,Schneider N,et al.Enzyme-catalyzed enantioselective hydrolysis of dihydrouracils as a route to enantiomerically pure β-amino acids[J].Catalysis,2011,1:1014 -1016.
[74]Bea H S,Park H J,Lee S H,et al.Kinetic resolution of aromatic β-amino acids by ω-transaminase[J].Chem Commun,2011,47:5894 -5896.
[75]Bach R D,Canepa C.Electronic factors influencing the decarboxylation of β-keto acids.Amodel enzyme study[J].J Org Chem,1996,61(18):6346 -6353.
[76]Kim J,Kyung D,Yun H,et al.Cloning and characterization of a novel β-transaminase from Mesorhizobium sp.strain LUK:A new biocatalyst for the synthesis of enantiomerically pure β-amino acids[J].Appl Environ Microbiol,2007,73(6),1772 -1782.
[77]Yun H,Lim S,Cho B K,et al.ω-Amino acid:Pyruvate transaminase from Alcaligenes denitrificans Y2k-2:A new catalyst for kinetic resolution of β-amino acids and amines[J].Appl Environ Microbiol,2004,70(4):2529-2534.
[78]Koszelewski D,Lavandera I,Clay D,et al.Formal asymmetric biocatalytic reductive amination[J].Angew Chem Int Ed,2008,47(48):9337 -9340.
[79]Hwang B Y,Kim B G.High-throughput screening method for the identification of active and enantioselective omega-transaminases[J].Enzyme and microbial technology,2004,34:429 -436.
[80]Truppo M D,Rozzell J D,Moore J C,et al.Rapid screening and scale-up of transaminase catalysed reactions[J].Org Biomol Chem,2009,7:395 -398.
[81]Schatzle S,Hohne M,Robins E,et al.Rapid and sensitive kinetic assay for characterization of ω-Transaminases[J].Anal Chem,2009,81(19):8244 -8248.
[82]Schatzle S,Hohne M,Robins K,et al.Conductometric method for the rapid characterization of the substrate specificity of amine-transaminases[J].Anal Chem,2010,82(5):2082 -2086.
[83]Savile C K,Janey J M,Mundorff E C,et al.Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture[J].Science,2010,329(5989):305 -309.
[84]Tasnádi G,ForróE,Fülop F.Improved enzymatic syntheses of valuable β-arylalkyl-β-amino acid enantiomers[J].Org Biomol Chem,2010,8:793 -799.
[85]Grayson J I,Roos J,Osswald S.Development of a commercial process for(S)-β-phenylalanine[J].Org Process Res Dev,2011,15(5):1201 -1206.
[86]Hee L J,Kiwon H,Jaiwook P,et al.Chemoenzymatic dynamic kinetic resolution of alcohols and amines[J].Eur J Org Chem,2010,6,999 -1015.
[87]Shakeri M,Engström K,Sandström A G,et al.Highly enantioselective resolution of β-amino esters by candida antarctica lipase A immobilized in mesocellular foam:Application to dynamic kinetic resolution[J].Chemcatchem,2010,2(5):534 -538.
[88]Wu B,Szymanski W,Wildeman S D,et al.Efficient tandem biocatalytic process for the kinetic resolution of aromatic β-amino acids[J].Adv Synth Catal,2010,352(9):1409 -1412.
[89]Szymanski W,Wu B,Weiner B,et al.Phenylalanine aminomutase-catalyzed addition of ammonia to substituted cinnamic acids:A route to enantiopure αand β-amino acids[J].J Org Chem,2009,74(23):9152-9157.
[90]Wu B,Szymanski W,Wybenga G G,et al.Mechanism-inspired engineeringofphenylalanine aminomutase for enhanced β-regioselective asymmetric amination of cinnamates[J].Angew Chem Int Ed,2012,51(2):482-486.
