美国药品不良反应监测体系简介及对我国的启示
2013-03-16刘花杨世民冯变玲
刘花 杨世民 冯变玲
(西安交通大学医学院药学系,陕西西安710061)
美国药品不良反应监测体系简介及对我国的启示
刘花 杨世民 冯变玲
(西安交通大学医学院药学系,陕西西安710061)
通过查阅资料和文献,从法律法规、组织机构、信息系统三方面介绍美国食品药品监督管理局的药品不良反应监测现状,为我国的药品不良反应监测工作提供建议。我国应进一步完善法律法规体系、组织机构和信息系统,提高药品不良反应监测水平。
药品不良反应;监测体系;启示
自从上世纪60年代“反应停事件”之后,药品安全问题尤其是上市后药品安全问题成为全球关注的焦点,世界各国纷纷采取措施解决药品不良反应(adverse drug reaction,ADR)问题。美国是较早开展ADR监测的国家,积累了丰富的经验,其所建立的ADR监测体系也是世界公认的最有效的体系之一,成为许多国家参照的标准。我国ADR监测工作始于上世纪80年代,经过多年的发展,取得了一定的成就,但是与发达国家相比还存有一定的差距。本文从美国ADR监测体系的法律法规体系、组织机构建设、技术体系方面进行分析介绍,以期为提高我国的ADR监测工作提供借鉴。
1 美国ADR监测体系介绍
1.1 法律法规体系
美国食品药品监督管理局(FDA)的主要任务就是执行美国国会制定的法律和该机构制定的法规条例来保证消费者的健康、安全。美国的ADR法律体系比较完备,包括法案、法规及指南性文件,共同确保ADR监测工作的高质量运行。
1.1.1 法案和法规
1.1.1.1 《联邦食品药品化妆品法案》(Federal Food,Drug,and Cosmetic Act,FDCA)[1]FDCA是美国食品和药品的基本法律。该法案由9部分组成,其中第5部分“药品和医疗机械”对于药品的监督管理作了细致的要求,并对ADR的相关工作作了明确的规定,例如Sec.355-1规定了风险评估和风险缓解策略(REMS),Sec.355b明确了不良事件报告等。
1.1.1.2 《联邦法典》(Code of Federal Regulations,CFR)[2]CFR是美国各种法律、法规的汇编,其中第21章“食品与药品”的部分条款对ADR作了详细的规定,例如 310.305部分介绍了关于未获得新药申请的已上市处方药的ADR的记录和报告,该部分明确了当ADR发生时需要向FDA提交报告的类型,而对于1938年之前上市并获得新药申请的药品不需要报告;314.80部分是关于上市后ADR的报告,该部分明确了获得新药申请的已上市药品当发生ADR时需要向FDA提交报告的类型;314.90部分是关于FDA对于批准已上市新药豁免的申请,并在314.50和314.81规定了豁免的要求;314.98部分是关于上市后报告,该部分明确了关于简化新药申请的药品发生ADR后报告和记录保存的要求。
1.1.1.3 《处方药使用者费用法案》(Prescription Drug User Fee Act,PDUFA)[3]1992年美国颁布了PDUFA,并于2007年完成第4次修订,该法案授权FDA向生产人用药品和生物制品的企业收取一定的费用。自从该法案获得通过,加快了FDA审批新药上市的速度。为了加强药品上市后的监管,2007年9月30日,总统签署《食品药品监督管理局2007修正法案》(FDAAA),标志其正式成为法律文件。在该法案的第9部分,首次提出“加强药品上市后的安全监管”,意味着FDA对于药品上市后的风险再评估获得了法律地位。
1.1.2 指南性文件 美国的指南性文件不具有法律的强制性,它只是代表了FDA对于目前监管方面存在的问题所采取的措施,仅作为参考的意见。因此,美国指南性的文件具有针对性强、更新速度快的特点。
2001年3月,FDA颁布了“人用药品和生物制品(包括疫苗)上市后安全报告”[4]的指南,该指南内容包括12部分,对于ADR的报告主体、报告内容、报告的种类、特殊报告情况以及如何提交报告等内容都作了明确的规定。2005年3月,FDA发布了关于药品风险管理方面的3个指南:《上市前风险评估》、《风险最小化执行方案的制定与应用》、《药物警戒规范与药物流行病学评估》。2009年10月1日,美国颁布了《关于推荐REMS,REMS评估以及推荐REMS的修改的格式和内容》的指南,旨在为药品生产企业提供以下指导:①推荐REMS的格式和内容;②评估的内容以及已批准REMS建议修改的内容;③REMS文件中标识符号的使用;④如何与FDA就REMS进行交流。2011年2月16日,美国又颁布了《使用电子医疗数据库开展和报告药物流行病学安全研究的良好实践》的指南,该指南包括指导药物流行病学安全研究的设计、分析和结果,以优化FDA对该类研究的审评方案和最终报告。该指南主要为药品生产企业和FDA的工作人员提供以下指导:①当药品生产企业向FDA提交药物流行病安全研究的方案和最终报告时,该指南能够为企业提供全程的指导以保证企业能够提交足够的信息通过审评;②为FDA审评人员提供一个审查和分析这类研究报告的框架;③为FDA提供这类研究实施过程中的全面指导。此外,FDA还颁布了《药品安全信息——FDA与公众的沟通》指南,对FDA如何向公众传递药品安全信息进行了说明。
1.2 组织体系
FDA是美国从事药品管理的最高执法机关。在美国,负责药品上市后安全的主要部门是药品审评研究中心(Center for Drug Evaluation and Research,CDER)和生物制品审评研究中心(Center for Biologics Evaluation and Research,CBER)。
1.2.1 CDER[5]CDER作为FDA的一部分,负责管理非处方药和处方药(包括生物制药和仿制药),通过确保药品的安全性和有效性来改善美国公众的生命健康。CDER由8个主要办公室组成,其中与ADR工作相关的办公室有中心主任办公室、执行办公室、医疗政策办公室、新药办公室以及监测与流行病学办公室,各办公室具体职责见表1。其中,监测与流行病学办公室承担了绝大部分的药品上市后监测工作,具体的部门设置及职责见表2。

表1 与ADR工作相关的办公室及其职责

表2 监测与流行病学办公室各部门职责
1.2.2 CBER CBER是FDA的一个中心,也是美国卫生人类服务部(HHS)的政府部门机构。它的职责是在适用的联邦法律下通过管理生物制品及其相关产品包括血液、疫苗、过敏原制剂、组织、细胞及基因治疗产品,来保护和提高公众健康。
1.2.3 药品安全监督委员会(Drug Safety Oversight Board,DSB)[6]DSB创建于2005年,并在2007年FDA修正案中得到规定。DSB由FDA的2个中心和8个其他的联邦政府机构组成。这些机构包括卫生保健研究和质量局(AHRQ)、疾病控制和预防中心(CDC)、医疗保险和医疗补助服务中心(CMS)、国防部(DOD)、卫生资源和服务管理局(HRSA)、印度卫生服务(IHS)、美国国立卫生研究院(NIH)、退伍军人事务部(VA)。DSB的一个重要作用就是帮助FDA评估他们的安全决策对于医疗保健系统以及联邦伙伴的影响。委员会由于其具有联邦医疗保健组织广泛的代表性,能够就一些重要的和经常出现的药品安全问题提供一些宝贵的意见,也可以使FDA能够获得有关药品安全问题的其他观点。DSB每月召开一次会议,举行论坛讨论如何处理潜在的药品安全问题。
1.3 技术体系
1.3.1 不良事件报告系统(Adverse Event Reporting System,AERS) AERS是一个旨在支持 FDA对药品和生物制品上市后监测计划的数据库,该数据库包含了FDA收集到的所有不良事件信息和用药错误信息。AERS的信息结构依据的是国际协调会议(ICH)E2B发布的国际安全报告指南,信息编码依据的是医学词典监管活动术语集中的条款。
目前美国有2个AERS:一个是针对于药品生产企业的强制报告系统;一个是针对于医疗专业人员和消费者的MedWatch系统。在美国,医疗专业人员和消费者报告不良事件和用药错误是自愿的,因此MedWatch系统是一个自愿报告系统。据统计,美国90%以上的不良事件报告都来自于药品生产企业。医疗专业人员和消费者报告时,既可以直接报告给FDA,也可以报告给药品生产企业,但是如果企业收到报告,就必须按照规定的要求提交给FDA。最后由FDA将直接和间接收到的报告统一录入到AERS。美国不良事件的报告主要有3种形式:网上报告、纸质报告和电话报告。
AERS对于FDA来说是一个有用的工具,利用该工具可以发现一个已上市药品存在的新的安全问题或是评价一个药品生产企业遵守报告管理及其对外界信息需求的反应情况。当一个药品获得FDA的批准后,CDER和CBER的临床审评专家就会对AERS中的报告进行评估以监测药品的安全性。一旦发现潜在的安全问题,就会进行进一步的评估。根据药品安全问题的评估情况,FDA就会采取一些措施提高药品的安全性,保护公众健康,例如更新药品标签信息,限制药品使用,将最新的安全信息与公众交流,或者罕见情况下召回药品。
1.3.2 “警戒倡议”(Sentinel Initiative)[7]2007年秋天,美国国会通过了FDAAA,授权FDA建立一个主动监测系统以监测药品和使用来自健康信息持有者的电子数据。“警戒倡议”就是对该授权所作出的回应。“警戒倡议”的公布通过了FDAAA,并于 2007年 9月成为法律。2008年5月,HHS和FDA宣布推出FDA“警戒倡议”。该项目旨在开发和运行一个警戒系统,该系统对于现在所使用的追踪管辖药品的AERS将是一个补充。它使得FDA能够积极查询多个医疗数据持有者(如电子健康档案系统,管理和保险索赔数据库),快速安全地评估医疗产品可能存在的安全问题,大大提高安全监管能力。目前,FDA建立警戒系统的试点方案包括:小警戒试点(Mini-Sentinel pilot)和联邦伙伴合作(Federal Partners’Collaboration,FPC)。
1.3.2.1 小警戒试点 它能够使FDA查询约60万患者持有的私人电子医疗数据(包括行政索赔和临床数据)。2009年,哈佛Pilgrim保健中心经FDA授 权 建 立 了 Mini-Sentinel Coordinating Center(MSCC),即警戒系统的缩小版。该系统使得FDA能够对各种科学方法和新的措施进行试验,以建立警戒系统。MSCC的运行过程见图1。
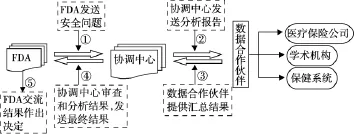
图1 MSCC安全问题评估过程
1.3.2.2 联邦伙伴合作 该试点包括医疗保险和医疗补助服务中心、退伍军人服务部以及国防部,它使FDA能够查询联邦电子医疗数据,包括来自电子健康档案系统的行政索赔数据。与小警戒试点不同的是,它不使用通用的数据模型,而是签订一个共同的主动监测协议,每个数据伙伴遵守协议用分析编码运行自己的数据库。
2 启示
2.1 完善法律法规体系,保障ADR工作各个环节的有效运行
完善的法律法规体系是ADR工作顺利开展的重要保障。未来我国法律法规体系的完善可从以下几个方面进行:①提高ADR监测工作的法律地位,确保ADR监测工作开展的强制性。目前,《药品不良反应报告和监测管理办法》可全面管理ADR监测,然而这只是一个部门规章,法律地位偏低。建议在《药品管理法》中明确ADR的法律地位。②完善现有的法律规章,明确ADR报告主体的职责和法律责任,细化ADR的奖罚政策,提高ADR报告的积极性。③为ADR报告主体制定各种标准操作规程和各项技术规范,形成统一的标准,指导ADR工作的开展,保证ADR监测工作的质量和规范性。④制定有关ADR信息公开交流方面的规范性文件,规范ADR传播行为,确保信息及时有效的传播,维护公众健康。
2.2 进一步完善ADR组织机构体系
完善的组织机构体系是ADR工作有序发展的基础,鉴于实际情况,我国仍保持ADR监测工作分级管理的模式。建议:①完善市县级ADR监测机构的建设,建立起广泛的ADR监测网络,确保ADR信息的全面收集。②统一全国省级ADR监测中心的建设模式,结束建设模式多样化的局面,保证全国ADR监测工作规范化。③进一步扩大和细化ADR监测机构的工作部门,分工细致,进一步明确各部门的职责,提高工作质量。④扩大工作人员数量,做到各项工作均有专人负责,保证各项工作的有效运行。⑤充分发挥专家咨询委员会的作用,明确职责和制定工作章程,实行规范化管理。
2.3 进一步完善信息系统
信息系统的应用给ADR监测工作提供了有力的技术支持,提高了工作效率。目前,随着药品安全问题的不断出现,我国的信息系统仍需改善:①开发消费者报告系统。与医疗专业人员相比,消费者专业知识缺乏,使用网上报告系统存在困难,建议开发适用于消费者的报告系统,报告系统应附有填写指南,指导消费者填写报告,提高ADR报告率。②提高系统的规范化。采用全球通用标准,对信息系统进行一些标准化的设置,例如上一项若填写不规范,将不能进行下一项的填写,提高ADR报告的规范性,不仅有利于提高工作人员的工作效率,也有利于国际间ADR信息的交流。③扩大信息系统的承载量。目前药物警戒是药品安全监管工作发展的国际化趋势,仅仅收集ADR信息已不能满足发展的需要,因此信息系统在保证满足ADR监测工作的基础上,可扩大信息收集范围,例如添加用药错误等信息。④加强与国内外其他数据库的连接,增加ADR信息来源,实现数据的共享,促进国际间的合作与交流,共同维护公众健康。
2.4 重视ADR信息的传播与交流
目前我国向公众传达ADR信息的途径主要是通过发布《药品不良反应信息通报》,交流形式比较单一,应用性不强。为增加公众对ADR信息的可及性,可利用一些常用的传播媒介,如监管部门与电视、广播等媒体合作,制作有关药品安全(包括 ADR)的栏目,根据实际情况,可以每周 1次的频率进行播放;或为了更及时有效地传达ADR信息,药监部门可与当地销售较好的报纸单位合作,刊登ADR信息等。此外,对于ADR信息的传播,应制定相关的行为规范,确保信息的真实、可靠、合法。
[1] FDA.Federal Food,Drug,and Cosmetic Act[EB/OL].(2011-5-12)[2012-6-13].http://www.fda.gov/RegulatoryInformation/Legislation/FederalFoodDrugandCosmeticActFDCAct/default.htm.
[2] FDA.Federal Regulations[EB/OL].(2009-7-27)[2012-6-13]. http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedicalProducts andTobacco/CDER/ucm169544.htm.
[3] FDA.Prescription Drug User Fee Act[EB/OL].(2012-5-30)[2012-6-13].http://www.fda.gov/ForIndustry/UserFees/PrescriptionDrugUserFee/default.htm.
[4] FDA.Center For Drug Evaluation and Research List of Guidance Documents[EB/OL].(2012-4-6)[2012-6-13].http://www.fda. gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/ Guidances/UCM079645.pdf.
[5]FDA.CDEROfficesandDivisions[EB/OL].(2012-3-21)[2012-6-13]. http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedical ProductsandTobacco/CDER/ucm075128.htm.
[6] FDA.Drug Safety Oversight Board[EB/OL].(2010-12-22)[2012-6-13].http://www.fda.gov/AboutFDA/CentersOffices/OfficeofMedical ProductsandTobacco/CDER/ucm082129.htm.
[7] FDA.The Sentinel Initiative[R].U.S.Food and Drug Administration,2010.
A Brief Introduction of the ADR Supervision System of FDA and Its Enlightenment to China
Liu Hua,Yang Shimin,Feng Bianling(Pharmacy Department of Medical College of Xi’an Jiaotong University,Shanxi Xi’an 710061,China)
This paper introduced the status quo of the ADR monitoring system of FDA including laws and regulations,organizations and information system based on literature review so as to provide a reference for the ADR supervision in China.It was suggested that further efforts should be made in China to improve the law and regulation system,organization and information system so that the ADR monitoring level can be raised.
Adverse Drug Reaction;Monitoring System;Enlightenment
2012-06-20)
10.3969/j.issn.1672-5433.2013.04.009
刘花,女,在读硕士。研究方向:药事管理。通讯作者E-mail:lxhzmei@163.com
